Simultaneous Quantitation of Buprenorphine and Its Metabolites Using LC–MS
Special Issues
An LC–MS method for simultaneous quantification of buprenorphine and three metabolites: norbuprenorphine, buprenorphine glucuronide, and norbuprenorphine glucuronide.
A liquid chromatography–mass spectrometry (LC–MS) method has been developed for simultaneous quantification of buprenorphine and its three metabolites, namely norbuprenorphine, buprenorphine glucuronide, and norbuprenorphine glucuronide. Chromatographic separation was achieved on a C18 column with a gradient of acetonitrile over ammonium acetate buffer (25 mM, pH 6.6). The method run time was 7.5 min. Quantification was performed by selected ion monitoring of [M+H]+ ions of norbuprenorphine glucuronide (590), norbuprenorphine (414), buprenorphine glucuronide (644), and buprenorphine (468). Naloxone (328) (328 ng/mL) was used as an internal standard. The samples were processed by protein precipitation and extraction recovery was ≥95% with minimal observed matrix effects (≤11%). Linear calibration curves were obtained over a range of 25 or 100 ng/mL to 4000 ng/mL, depending on the analyte. The lower limit of quantitation for buprenorphine and norbuprenorphine was 25 ng/mL and for buprenorphine glucuronide and norbuprenorphine glucuronide it was 100 ng/mL. The intraday and interday precision and accuracy determinations were <15% coefficient of variation and ≤15% bias, respectively. The method was successfully applied to in vitro enzymatic metabolism studies of buprenorphine.
Buprenorphine (BUP) is a partial µ-opioid receptor agonist and κ-opioid receptor antagonist that displays 25–40-fold potent analgesic activity than morphine (1). It appears to display lower potential for addiction and lesser respiratory depression compared to morphine (2). While BUP displays good oral absorption in the gastrointestinal tract, it suffers from very low oral bioavailability because of extensive presystemic metabolism in the gut and the liver (3,4). The majority of BUP metabolism is through oxidation by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C8 to form norbuprenorphine (NBUP) (Figure 1) (5–7). In addition, BUP and NBUP are also metabolized by phase II pathways such as glucuronidation to produce BUP-glucuronide (BUPG) and NBUP-glucuronide (NBUPG), respectively (Figure 1) (8–11). BUP in combination with naloxone (NX) (Suboxone, Indivior UK Limited) was approved in the United States for the treatment of opioid dependence in 2002.


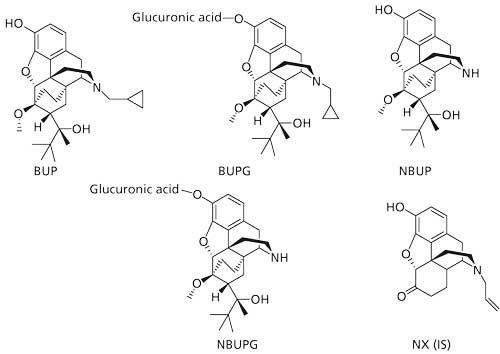
Figure 1: Structures of analytes and internal standard.
Early analytical methods for quantitation of BUP included analysis by high performance liquid chromatography (HPLC) coupled to fluorescence detection (12), ultraviolet (UV) detection (13), or use of gas chromatography (GC) (14). In addition, several liquid chromatography–mass spectrometry (LC–MS) methods have also been reported for quantitation of BUP in plasma (15–17), serum (18), whole blood (19), and urine (16,20,21). In the previous reports, reversed-phase HPLC has been extensively used for chromatographic separation of BUP and its metabolites (16,18–25). Previously developed LC–MS-based methods for quantifying BUP and its metabolites have been reviewed elsewhere (26,27).
A benchtop mass spectrometer similar to a single-quadrupole detector was used in this study. While several methods developed in the past have used reversed-phase HPLC for quantitation of BUP and its metabolites (as stated above), reports for quantitation of BUP and its metabolites using reversed-phase HPLC coupled to this detector have not been reported. To the best of our knowledge, we report for the first time the use of reversed-phase HPLC coupled to this benchtop MS detector for quantifying BUP and its three metabolites: NBUP, BUPG, and NBUPG. We believe that as this benchtop MS detector grows in popularity it would be worthwhile and beneficial to report a validated LC–MS method for BUP and its metabolites using it.
The aim of the present study was to develop and validate a LC–MS method for simultaneous quantitation of BUP, NBUP, BUPG, and NBUPG using reversed-phase HPLC coupled to a benchtop mass spectrometer. The method was successfully developed and validated and had a run time of 7.5 min. Further, the method was successfully applied to in vitro microsomal metabolic studies of BUP.
Experimental
Materials
LC-grade solvents such as acetonitrile were purchased from Pharmaco and Aaper. Reference standard solutions of BUP, NBUP, BUPG, and NBUPG were purchased from Cerilliant. NX and BUP for metabolism studies were purchased from Medisca. Ammonium acetate was obtained from EMD Chemicals Ltd. A 100 mm x 4.6 mm, 3-µm dp C18 Alltech Alltima HP column for LC–MS analysis was obtained from Grace Discovery Sciences. Human liver microsomes (HLM) for metabolism studies were obtained from Xenotech. NADPH and MgCl2 were purchased from Akron Biotechnology LLC and Fisher Scientific, respectively. Bovine serum albumin (BSA) was purchased from Gemini Bio-Products.
Preparation of Calibration Standards and Quality Control Samples
Commercially available stocks were obtained for NBUP, BUPG, and NBUPG at 100 µg/mL in methanol and BUP at 1 mg/mL in methanol. The stocks were stored at -20 °C until the day of the experiment. A solution of 0.2 mg/mL of HLM in 0.1 M potassium phosphate buffer with 0.05% BSA was used as a blank matrix. Blank matrix was used as a diluent for preparing calibration standards of appropriate concentrations from the methanolic stocks. The calibration standards for BUP and NBUP were 25, 50, 100, 200, 500, 1000, 2000, and 4000 ng/mL and 100, 200, 500, 1000, 2000, and 4000 ng/mL for BUPG and NBUPG. The quality controls (QC) were also prepared similar to the calibration standard solutions. Three levels of QCs with final concentration of 100 (low-QC), 2000 (mid-QC), and 4000 ng/mL (high-QC) for NBUP and BUP and 500 (low-QC), 2000 (mid-QC), and 4000 ng/mL (high-QC) for BUPG and NBUPG were prepared.
Sample Preparation
The samples from the enzymatic metabolism studies of BUP (100 µL) were mixed with an equal volume of acetonitrile (100 µL) containing NX (328 ng/mL) as the internal standard. The samples were thoroughly vortexed to precipitate protein. Subsequently, the samples were centrifuged to separate the supernatant. A part of the supernatant (100 µL) was mixed with blank potassium phosphate buffer (no BSA) (100 µL) and the resulting solution was injected (50 µL) into the LC–MS. The samples were maintained at 5 °C during the analysis.
LC–MS Method Development
HPLC Conditions
The LC system consisted of a model 2695 HPLC system (Waters). All four of the analytes (BUP, NBUP, BUPG, and NBUPG) were retained on the C18 column. The LC method included a gradient of 99% A (10% acetonitrile, 90% aqueous, 25 mM ammonium acetate, pH 6.6, 5 µL glacial acetic acid per liter) and 1% B (neat acetonitrile) for the initial first min (0–1 min) followed by a gradient from 1% to 50% B over 1.5 min (from 1 to 2.5 min). Subsequently, the gradient was ramped from 50% to 90% B over 0.5 min (from 2.5 to 3 min) and further, 90% B was maintained for 3 min (from 3 to 6 min). This was followed by a re-equilibration to 1% B for 1.5 min (until 7.5 min). The flow rate was constant at 1 mL/min. The column temperature during the analysis was 30 °C. The eluent was diverted to waste for the initial 3 min. The total run time was 7.5 min (Table I).


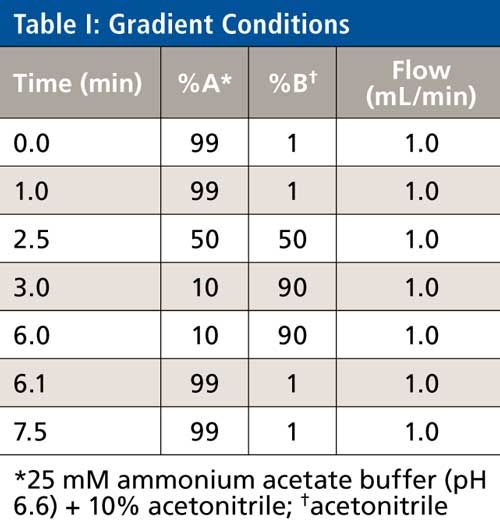
MS Conditions
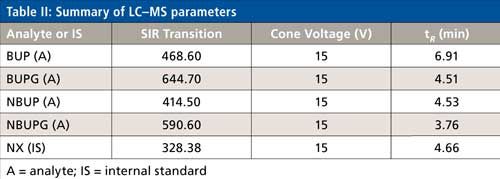
The MS system was an Acquity QDa mass spectrometer (Waters), which is similar to a single-quadrupole system. The eluent from the analytical column was diverted to the waste for initial 3 min. The eluent from 3 to 7.5 min was injected into the mass spectrometer. The capillary positive voltage was set at 0.8 kV and the probe temperature was 600 °C. Instrument control, acquisition, and data processing were performed using Empower 3 software (Waters). The single ion recording (SIR) and cone voltages for each analyte are noted in Table II. The source, ion block, and sample cone were cleaned weekly.


LC–MS Method Validation
Linearity and Lower Limit of Quantitation
Each calibration curve consisted of eight different levels of concentrations of calibration standards in duplicate for BUP and NBUP and six levels for BUPG and NBUPG. The calibration curves were fitted to nonlinear regression using a quadratic equation and 1/y2 as the weighting factor. The regression equation was:


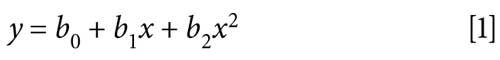
The calibration parameters were calculated by using GraphPad Prism 5 software and the nonlinear regression equation was used to determine the sample concentrations. The lowest concentration of the calibration curves was the lower limit of quantitation (LLOQ) for all four analytes.
Precision, Accuracy, Extraction Recovery, Matrix Effects, and Autosampler Stability
The method was evaluated for intraday precision and accuracy for all four of the analytes by analyzing at least five replicates at three concentration levels of QC samples and at LLOQ as well and thereby calculating the percent coefficient of variation (%CV) and percent bias, respectively. Interday precision and accuracy was evaluated by calculating %CV and %bias, respectively, and analyzing samples at three levels of QCs that were run on four days. Interday precision and accuracy was also calculated at LLOQ on three days. The extraction recovery of the analyte was also measured at three concentration levels of QC sample in at least triplicates by comparing mean peak area obtained from blank matrix solutions spiked with the analyte before protein precipitation to the mean peak area obtained by spiking with analyte after protein precipitation. The matrix effect was also calculated at the three levels of QC sample in at least triplicate by comparing the mean peak area obtained from post protein precipitation spiked blank matrix solutions to that with the mean peak area of external analyte solution. Carryover was evaluated by injecting high QC sample followed by a blank solvent used for sample preparation. The autosampler stability of the analytes was assessed following 14 h of storage in the autosampler at 5 °C. The analytes were evaluated at four levels of concentrations: 100, 500, 2000, and 4000 ng/mL followed by a calculation of %CV.
Enzymatic Study
The reaction mixture comprised of 0.2 mg/mL HLM, 1 mM NADPH, 2.5 mM UDPGA, 12.5 mM MgCl2, 8.1 mM saccharolactone, 31.25 µg/mL alamethicin, 8 µM BUP, and 0.1 M potassium phosphate buffer (pH 7.4) containing 0.05% (w/v) BSA. The microsomes were preactivated by incubation with alamethicin on ice for 20 min. The reaction was initiated by adding UDPGA and NADPH followed by incubation at 37 °C for 30 min. An equal volume of cold acetonitrile containing the internal standard (328 ng/mL NX) was used to stop the reactions. Subsequently, protein precipitate was separated from the supernatant by centrifugation and the supernatant was stored at -20 °C until further analysis. The incubation was performed in triplicates.
Results
Optimization of LC–MS Conditions
The chromatographic conditions were optimized on an Alltech Alltima HP C18 column using a mobile phase containing acetonitrile and 25 mM ammonium acetate buffer. All of the analytes were eluted in the 3.6–7 min range. The chromatographic gradient was optimized such that there was sufficient time for the buffer salts to be eluted to the solvent waste (initial 3 min of the run time) followed by a sequential elution of all four analytes including the internal standard until 7 min. These chromatographic gradient conditions produced a quantifiable and reproducible peak response for all the analytes and internal standard. The selected ion recording (SIR) used for each analyte was as follows: BUP = 468.6, NBUP = 414.5, BUPG = 644.7, NBUPG = 590.6, and NX = 328.3 (Table II and Figure 2a–e). Blank samples containing matrix with and without an internal standard did not display background peaks for all four analytes. Baseline resolution was achieved between all the analytes except for between NBUP and BUPG. Although baseline resolution was not considered necessary for quantification of metabolites because of mass selective detection, we also performed a short study to verify whether there was any bleeding of NBUP into the BUPG channel and thus, whether NBUP contributes to the chromatographic peak area of BUPG. We were prompted to perform this study because of the presence of a mass peak of NBUP (m/z = 414) following integration of the chromatographic BUPG peak in the BUPG channel. To investigate this m/z 414 peak, a high concentration solution (4000 ng/mL) of NBUP was injected into the LC–MS and the BUPG channel was monitored for the presence of a NBUP chromatographic peak. However, no peak was observed. Further, a low-concentration solution of BUPG (400 ng/mL) along with and without a high-concentration solution (4000 ng/mL) of NBUP were injected onto the LC–MS system. The chromatographic peak areas of BUPG peaks were similar (less than 5% difference) with and without the presence of NBUP, indicating that NBUP contributes minimally to the chromatographic peak area of BUPG.


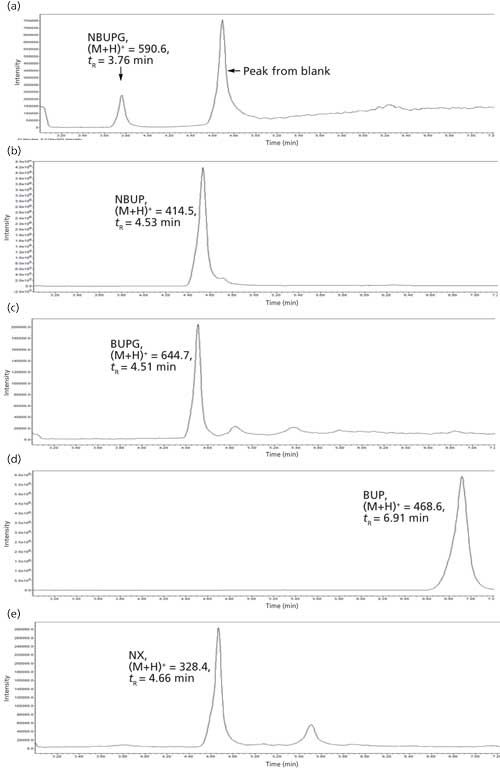
Figure 2: Representative chromatograms of analytes and internal standard.
Selection of an Internal Standard
The ideal internal standard for LC–MS analysis is the isotopically labeled analyte. While the use of an isotopically labeled analyte NBUP-d3 was attempted, a spillover was observed into the NBUP channel, possibly because of an isotopic effect. While the spillover was minimal, NX also displayed a retention time in the 3.6–7 min time period and displayed good sensitivity. Hence, an NX concentration of 328 ng/mL that produced a quantifiable peak response was used as the internal standard for the analysis.
LC–MS Method Validation
Linearity and LLOQ
The calibration curves for the BUP and NBUP displayed good linearity over the concentration range of 25–4000 ng/mL and 100–4000 ng/mL for BUPG and NBUPG. The correlation coefficients (r2) for all calibration curves were greater than 0.99, except one calibration curve for BUP that displayed a correlation coefficient of 0.985. All the calibration curves were run in duplicate for all the analytes. The calibration curves for NBUP, BUPG, NBUPG, and BUP were fitted by nonlinear regression to a quadratic equation. The LLOQ of NBUP and BUP is 25 ng/mL and that for NBUPG and BUPG is 100 ng/mL. The %CV and %bias for LLOQ was less than 20%.
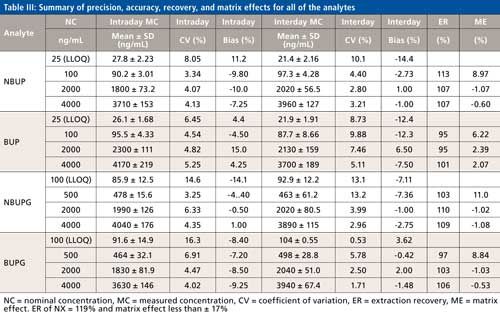
Precision, Accuracy, Extraction Recovery, Matrix Effects, and Autosampler Stability
The precision and accuracy for all four analytes were evaluated at three levels of QC and also at the LLOQ. For all four analytes, while the intraday and interday precision ranged from 3.25% to 16.3% and 0.53% to 13.2%, respectively (Table III), the intra- and interday accuracy ranged from -14.1% to 15.0% and -14.4% to 6.50%, respectively (Table III). The matrix effect for all the analytes was less than or equal to 11% (Table III). The recoveries of all analytes following protein precipitation ranged from 95% to 113% (Table III). All of the analytes displayed a minimal carryover (<1%). The %CV determined at the four concentration levels in the autosampler stability test was less than 15% for all of the analytes.


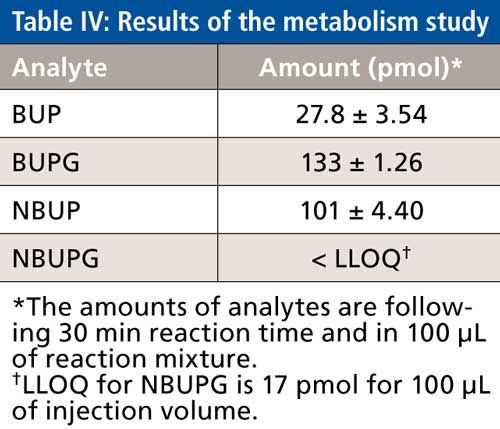
Metabolism Study of BUP
BUP concentrations at the end of 30 min were consistent with almost complete conversion to its metabolites. Of the BUP metabolites, the highest concentrations of BUPG were observed followed by NBUP whereas NBUPG was below the LLOQ (Table IV). Successful quantitation of BUP and its metabolites BUPG and NBUP was performed using this analytical method.


Conclusions
An LC–MS method was developed and validated for the simultaneous quantitative determination of BUP and its three metabolites (BUPG, NBUP, and NBUPG) with a run time of 7.5 min. The method displayed good linearity and acceptable inter- and intraday precision and accuracy. All of the analytes displayed more than 95% extraction recoveries and matrix effects that were less than or equal to 11%. This method was further successfully applied for the analysis of BUP and its metabolites from microsomal metabolism studies. By diverting to waste for the initial part of the run, no sample extraction was necessary for mass detection, and equipment maintenance was minimal. We believe this method would serve as a good reference for laboratories using the benchtop MS detector.
References
- A. Cowan, J.W. Lewis, and I.R. Macfarlane, Br. J. Pharmacol. 60, 537–545 (1977).
- M.R. Lofwall and S.L. Walsh, J. Addict. Med. 8, 315–326 (2014).
- M. Mistry and J. Houston, Drug Metab. and Dispos. 15, 710–717 (1987).
- D. Brewster, M. Humphrey, and M. McLeavey, J. Pharm. Pharmacol. 33, 500–506 (1981).
- C. Iribarne, D. Picart, Y. Dreano, J.P. Bail, and F. Berthou, Life Sci.60, 1953–1964 (1997).
- K. Kobayashi, T. Yamamoto, K. Chiba, M. Tani, N. Shimada, T. Ishizaki, and Y. Kuroiwa, Drug Metab. Dispos. 26, 818–821 (1998).
- N. Picard, T. Cresteil, N. Djebli, and P. Marquet, Drug Metab. Dispos.33, 689–695 (2005).
- S. Oechsler and G. Skopp, Int. J. Legal Med. 124, 187–194 (2010).
- H.E. Cubitt, J.B. Houston, and A. Galetin, Pharm. Res. 26, 1073–1083 (2009).
- K.A. Seo, H.J. Kim, E.S. Jeong, N. Abdalla, C.S. Choi, D.H. Kim, and J.G. Shin, Drug Metab. Dispos. 42, 1803–1810 (2014).
- P.J. Kilford, R. Stringer, B. Sohal, J.B. Houston, and A. Galetin, Drug Metab. Dispos.37, 82–89 (2009).
- S.T. Ho, J.J. Wang, W. Ho, and O.Y. Hu, J. Chromatogr. 570, 339–350 (1991).
- L.P. Hackett, L.J. Dusci, K.F. Ilett, S.S. Seow, and A.J. Quigley, J. Chromatogr.374, 400–404 (1986).
- Y. Blom, U. Bondesson, and E. Anggard, J. Chromatogr. 338, 89–98 (1985).
- C.M. Murphy and M.A. Huestis, J. Mass Spectrom. 40, 70–74 (2005).
- K.J. Regina and E.D. Kharasch, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.939, 23–31 (2013).
- Y. Liu, X. Li, A. Xu, A.F. Nasser, and C. Heidbreder, J. Pharm. Biomed. Anal.120, 142–152 (2016).
- M. Scislowski, W. Piekoszewski, A. Kamenczak, and E. Florek, J. Anal. Toxicol. 29, 249–253 (2005).
- H. Hoja, P. Marquet, B. Verneuil, H. Lotfi, J.L. Dupuy, and G. Lachatre, J. Anal. Toxicol.21, 160–165 (1997).
- G.A. McMillin, R. Davis, H. Carlisle, C. Clark, S.J. Marin, and D.E. Moody, J. Anal. Toxicol. 36, 81–87 (2012).
- A.C. Liu, T.Y. Lin, L.W. Su, and M.R. Fuh, Talanta75, 198–204 (2008).
- A. Tracqui, P. Kintz, and P. Mangin, J. Forensic Sci. 42, 111–114 (1997).
- A. Polettini and M.A. Huestis, J. Chromatogr. B Biomed. Sci. Appl.754, 447–459 (2001).
- A. Ceccato, R. Klinkenberg, P. Hubert, and B. Streel, J. Pharm. Biomed. Anal. 32, 619–631 (2003).
- S. Hegstad, H.Z. Khiabani, E.L. Oiestad, T. Berg, and A.S. Christophersen, J. Anal. Toxicol. 31, 214–219 (2007).
- M. Barroso, E. Gallardo, D.N. Vieira, J.A. Queiroz, and M. Lopez-Rivadulla, Anal. Bioanal. Chem.400, 1665–1690 (2011).
- D. French, Bioanalysis5, 2803–2820 (2013).
Anand A. Joshi, Neha V. Maharao, and Phillip M. Gerk are with the Department of Pharmaceutics in the School of Pharmacy at Virginia Commonwealth University in Richmond, Virginia. Direct correspondence to: pmgerk@vcu.edu














New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.


Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

