The Role of Injection Solvents
LCGC Asia Pacific
The solvent chosen for injection can affect the apearance of chromatographic peaks.
The solvent chosen for injection can affect the appearance of chromatographic peaks.
A recent observation in the laboratory of one of the authors (B.A.) of this column highlights a problem that has been mentioned in "LC Troubleshooting" discussions before, but its importance justifies additional discussion about the selection of the injection solvent to use with a liquid chromatography (LC) method. In this particular case, it was observed that the peak areas were constant with two different injection solvents, but the peak heights were not. Similar results were observed with methods for two different compounds (A and B). Let's see what the likely cause is for this problem.
Background
We can't share the complete details of the methods because they are proprietary, but the key elements follow. A 10 ppm concentration of sample is prepared in distilled water or in 100% methanol, and 20 μL of this solution is injected. A 250 mm × 4.6 mm C18 column packed with 5-μm diameter particles is used with UV absorbance detection at 210 nm. For compound A (method A), a mobile phase of 60:40 methanol–water is used at a flow rate of 1.1 mL/min; for compound B (method B), the mobile phase is 70:30 (methanol–water) with a flow rate of 1.4 mL/min. The data are summarized in Table 1. Both compounds (A and B) are well retained with k-values of approximately 2.6 in each case. You can see that the peak area differences between injection in methanol and water are ≤0.1% for both methods. However, the peak heights are approximately 30% larger for the water injections.



Table 1: Chromatographic results for two methods A and B.
Effect of Injection Solvent
The chromatograms of Figure 1 illustrate the influence of the injection solvent on the appearance of peaks in the chromatogram for another sample (1). In each case, the reversed-phase column was operated with an 18% acetonitrile–buffer mobile phase, where the buffer was 81:1 water–acetic acid. Injection volumes of 30 μL were made in various injection solvent compositions.

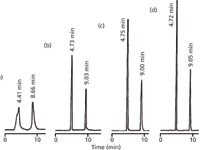
Figure 1: Chromatograms for caffeine (first peak of each chromatogram) and salicylamide (second peak). Injections of 30 μL of ~1 mg/mL of each analyte dissolved in (a) acetonitrile, (b) mobile phase, (c) 81:1 waterâacetic acid, and (d) water. Column: 250 mm à 4 mm, 10-μm dp; mobile phase: 18:81:1 acetonitrileâwaterâacetic acid; flow rate: 1 mL/min. Adapted from reference 1.
First, consider the cases where the injection solvent is no stronger than the mobile phase [Figures 1(b)–1(d)]. In each case, the retention times of each peak are approximately the same. Although we don't have data for the peak areas in each case, we'll assume they are the same, because the same volume of the sample at the same concentration was used in each case. Note, however, that the peak heights differ. Water is the weak mobile-phase solvent in this example, and the less water that is present in the injection solvent, the shorter the peak heights are. These observations are consistent with the problem presented at the beginning, where peak areas and retention times were constant, but peak heights dropped when less water was used in the injection solvent.
Next, notice what happens when the sample of Figure 1(a) was injected in 30 μL of 100% acetonitrile — the peaks are broader, distorted and at shorter retention times. This example continues the pattern of Figures 1(b)–1(d), where the peaks are broader and shorter as less water is used in the injection solvent.
Conceptually, What's Going On?
Let's think on a conceptual basis about what is going on with the various examples of Figure 1. If the sample is injected with mobile phase as the injection solvent, it does not have to undergo any chemical changes, so analyte molecules should immediately begin to interact with the stationary phase in the column and undergo normal retention behaviour. However, if the injection volume is large enough, you can imagine that some band broadening will take place during the injection. For example, if the injection is extremely large, such as 1 mL, the first sample molecules will travel a significant way through the column before the last of the sample molecules arrive at the inlet. The distance between the first and last sample molecules in a given band determine the peak width, so a broad peak would be observed. At the other extreme, an infinitely small injection would have none of this injection-related broadening, so the peak would be much narrower. As we'll see later, we generally can tolerate an injection volume of approximately 15% of the peak volume of the first peak of interest before we find the injection-related band broadening objectionable.
When the injection solvent is changed, we have another variable in addition to the injection volume mentioned above. In this case, the injected solvent plug must be diluted to the same composition as the mobile phase before "normal" retention can occur. If we think of the injection plug as a ball of solvent at the inlet of the column, it will shrink as it travels through the column, with the outside edges being diluted to the mobile-phase concentration before the centre of the ball. While they are inside the ball of the injection solvent, molecules will travel down the column as if that solvent was the mobile phase, whereas those diluted into the mobile phase will now travel at the normal rate in the mobile phase. So if the injection solvent is stronger than the mobile phase, it will tend to sweep analyte molecules through the column more quickly than normal. Because some analyte molecules travel more quickly (in the middle of the injection plug) than others (on the outer edges), injection in a solvent stronger than the mobile phase tends to smear the sample along the column, causing peak distortion. On the other hand, if the injection solvent is weaker than the mobile phase, analyte molecules inside the injection plug will move more slowly than normal, which tends to concentrate them at the column inlet. This is often referred to as on-column concentration and is a technique that can be used to inject large volumes of dilute samples without excessive band broadening. And, as you might expect, the influence of the injection solvent depends on both its volume and how much its composition differs from the mobile phase. A small volume of a strong solvent will be less detrimental than a large volume of the same injection solvent, and at a small enough volume, even a very strong injection solvent will get diluted very quickly, so it will be of little consequence.
How Much Is Too Much?
It can be shown (see discussion of reference 2) that if you want to have <1% loss in resolution because of injectionrelated peak broadening, the injection volume should be <15% of the peak volume of the first peak of interest if mobile phase is used as the injection solvent. If you are willing to tolerate a 10% loss in resolution, you can increase this to <40% of the peak volume. You can determine the peak volume of a peak by measuring its baseline width (or half-height width × 1.7) and multiplying by the flow rate. Alternatively, you can get a good estimate of the peak volume with a few simple calculations that follow. We think it is a good idea to make this calculation and compare the results with the actual measurements as a doublecheck to make sure there isn't excessive peak broadening because of other causes, such as a bad column.
We can use the data of Figure 1 as an example. The expected width can be estimated from the column plate number, N:


where tR is the retention time and w is the peak width at baseline between tangents drawn to the sides of the peak. Equation 1 can be rearranged to


Now we need a value of N, which for real samples under practical separation conditions can be estimated as


where L is the column length (in millimetres) and dp is the particle diameter (in micrometres). The column of Figure 1 is 250 mm long packed with 10-μm particles, so N ≈ 7500. Injected in the weakest solvent, water [Figure 1(d)], retention times are 4.72 and 9.05 min. With equation 2, this translates to expected peak widths of 0.0545 and 0.1045 min, respectively. (The usual disclaimer applies here: Values have been rounded for convenience, so if you try to reproduce these calculations, your results may vary somewhat.) Multiply by the flow rate (1 mL/min) to convert this to peak volumes of 55 and 105 μL.
If we use the <15% rule mentioned at the beginning of this section, maximum recommended injection volumes in mobile phase are then 0.15 × 55 = 8 μL and 0.15 × 105 = 16 μL. If we are interested in the first peak, this will be the limiting case, so injection of 5–10 μL in mobile phase would be expected to cause less than 1% loss in resolution. If we are more liberal with the tolerance for loss of resolution, as might be justified in the case of Figure 1, where resolution >>2, the <40% rule should be adequate, or 0.4 × 55 = 22 μL. Similar calculations can be performed for the data of Table 1 and we would see that the <15% rule would allow approximately 10-μL injections and the <40% rule would allow approximately 30-μL injections.
Does This Make Sense?
Now, the final test has to do with whether or not all these estimates line up with what is observed in reality. First, let's consider the data of Figure 1. Resolution is directly related to peak width, so a 1% or 10% loss in resolution would correspond to a 1% or 10% increase in peak width, respectively. Also, any increase in peak width should give a corresponding decrease in peak height, so we can indirectly measure any loss of resolution by a reduction in peak height. The 30 μL injections of Figure 1 are approximately 50% larger than the 22-μL injection volume estimated above for a 10% loss in resolution. You can see that the peak heights for injection of 30 μL of sample in mobile phase [Figure 1(b)] are 60–70% of those when 30 μL of water is injected [Figure 1(d)]. So some broadening is occurring, as expected –partly because of the injection in mobile phase rather than a weaker solvent and partly because of the larger than recommended injection volume. Note that in all cases where the mobile phase or weaker injection solvent was used [Figures 1(b)–1(d)], the retention times were unchanged (within normally expected sample-to-sample variability).
Contrast the results of Figures 1(b)–1(d) with those of Figure 1(a). In the case of Figure 1(a), both too large a sample was injected and too strong an injection solvent was used. This resulted in two problems. First, the peaks broadened unacceptably, especially for the first peak, which is severely distorted. Second, some of the sample molecules are swept down the column during the injectionsolvent dilution process, giving shorter retention times. These problems could be solved by injecting a much smaller sample volume or using a more dilute injection solvent.
The data of Table 1 show the same pattern, but less severely. This is because of at least two factors. First, the injected volumes (20 μL) are less than those of the <40% rule, and the difference between the injection solvent strength and the mobile phase is less. For Figure 1(a), 30 μL of acetonitrile is injected into an 18% acetonitrile mobile phase, whereas in Table 1, 20 μL of methanol is injected in a 60% or 70% methanol mobile phase.
Conclusions
We have seen that the combination of injection solvent strength and injection solvent volume must be chosen carefully to avoid unwanted band broadening and the resulting loss of resolution. In the examples of Table 1 and Figures 1(b)–1(d), the injection conditions were responsible for some loss in resolution (measured indirectly from loss in peak height), but no change in retention was observed. The peaks of Figure 1(a), however, were distorted and lost retention because too much of too strong a solvent was injected.
The <15% and <40% rules discussed above are guidelines that seem to work fairly well. But, as with all guidelines, it is a good idea to give the actual results a reality test. One way was discussed here, where estimates of peak volumes and injection conditions were compared with actual experimental conditions. Another technique is simply empirical – try increasing or decreasing the proposed injection volume by twofold. In the same manner, injection solvent concentrations that are closer to the mobile phase than the 100% strong or weak solvent conditions used in Table 1 and Figure 1(a) would be expected to have a less detrimental result. For example, injection in 70% methanol into a 60% methanol mobile phase would be expected to be less of a problem than an injection solvent of 100% methanol. If significant differences in the chromatograms are observed, you should adjust the conditions so that you are not operating too close to the reliability limits of the method.
Bandar Alsehli is a PhD student in the laboratory of Dr Hugh Flowers at Glasgow University in Scotland.
John W. Dolan is vice president of LC Resources, Walnut Creek, California, USA. He is also a member of LCGC Asia Pacific's editorial advisory board. Direct correspondence about this column should go to "LC Troubleshooting", LCGC Asia Pacific, 4A Bridgegate Pavilion, Chester Business Park, Wrexham Road, Chester, CH4 9QH, UK, or e-mail the editor, Alasdair Matheson, at amatheson@advanstar.com
References
(1) T.-L. Ng and S. Ng, J. Chromatogr. A 329, 13–24 (1985).
(2) L.R. Snyder, J.J. Kirkland and J.W. Dolan, Introduction to Modern Liquid Chromatography, 3rd Ed. (Wiley, Hoboken, New Jersey, 2010), section 2.6.1.

















New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

