The Role of Chromatography Data Systems in Fraud and Falsification


Chromatography data systems (CDSs) have had a starring role in many regulatory citations involving falsification and fraud in analytical laboratories regulated by Good Manufacturing Practices (GMP). In this instalment of "Questions of Quality" we will examine the citations and identify the technical and procedural controls required to ensure data integrity within these systems. Although focused primarily on the pharmaceutical industry, the principles described here are applicable to all laboratories working to established quality standards.
Chromatography data systems (CDSs) have had a starring role in many regulatory citations involving falsification and fraud in analytical laboratories regulated by Good Manufacturing Practices (GMP). In this instalment of "Questions of Quality" we will examine the citations and identify the technical and procedural controls required to ensure data integrity within these systems. Although focused primarily on the pharmaceutical industry, the principles described here are applicable to all laboratories working to established quality standards.
There has been a growing increase in the number of laboratories found guilty of falsification and fraud when chromatography data systems (CDSs) operating in Good Manufacturing Practices (GMP) regulated laboratories have been inspected by the United States and the European regulatory agencies. The inspection focus has changed: Instead of wading through reams of paper printouts, the inspection now reviews the electronic records in the CDS. The reason for this change in focus initially began with the Able Laboratories fraud case in 2005 (1). Up until this point, the company had had multiple US Food and Drug Administration (FDA) inspections with no non-compliances. That was until a whistle blower called the local agency office to raise concerns about the working practices that were not entirely compliant with the regulations.
Some of the innovative analytical techniques employed were a combination of:
- Copy and pasting chromatograms from passing batches to failing ones;
- Extensive reintegration of chromatograms to ensure passing results;
- Adjustments of weights, purity factors, and calculations to ensure acceptable results.
This was how an original result of 29%, which would fail a specification of >85%, was falsified to a passing result of 91% (2). At the heart of the fraud was a CDS, which, when investigated by the FDA, had an audit trail that identified the individuals responsible for the falsification of data. Identification of the problems in the laboratory led to the closing of the company in 2005 (2) and the criminal prosecution of four individuals in 2007 (3).
The Able Laboratories fraud case has led to a review of the FDA's inspection approach. This has resulted in the rewrite of Compliance Programme Guide (CPG) 7346.832 for Pre-Approval Inspections (PAI). There are three objectives contained within the guide, one of which is objective 3 — the data integrity audit — that is focused on the laboratory (4). Before this came into effect in May 2012, FDA inspectors were given training in data integrity, including training by Monica Cahilly from Green Mountain Quality Assurance. The training focused on the computer system and the records it contains rather than the paper output. This focus on the CDS in regulated GMP laboratories has seen an increasing number of warning letter citations in the last 2 to 3 years.
In Europe, the UK's MHRA (Medicine's and Healthcare products Regulatory Agency) announced in December 2013 (5) that they expected companies to focus on data integrity in their self-inspections under EU GMP Chapter 9 (6). This applies not only within an organization but also in their supply chain. The website also helpfully supplies an e-mail address for whistle blowers.
MHRA has recently written to all major CDS suppliers requesting copies of their software so that they can understand how each individual software package works. In early April 2014, the MHRA inspectors, together with inspectors from other European regulatory agencies, had the same data integrity training as the FDA. Life is getting interesting for chromatographers in regulated laboratories!
FDA Warning Letters and European Non-Compliances for CDS
This column will focus on chromatography data systems and the role that they play in fraud and falsification cases, as documented in FDA warning letters (7) and European Medicines Agency GMP non-compliances (8). In general, the FDA warning letters tend to contain more detail and be far more critical of non-compliances.
When researching the warning letters for this column I have been very selective. I want to focus on the use of CDSs in falsification rather than surrounding areas of chromatographic analysis such as training, poor sampling, or not investigating out of specification results. It is the use, or rather misuse, of the CDS informatics software together with the operating system and utility software that is the focus of this column. In addition, when reviewing the warning letters on both the European Medicines Agency (EMA) and FDA websites, the search tools are rather primitive. Rather than list all relevant companies and go through each warning letter in detail, a summary of most of the regulatory citations is presented in Figure 1. I will discuss each area in more detail and, where appropriate, I will reference an individual warning letter or non-compliance where it serves as a good example of bad practice or where I want to make a specific point.


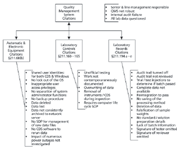
Figure 1: Collated and classified CDS citations from FDA warning letters involving a CDS.
The EMA non-compliances tend to be very general compared with the FDA, which cite against a specific section of the US GMP regulations, and therefore Figure 1 is biased towards the US regulations (9). Where there is no GMP regulatory citation given I have assigned what I think is most appropriate; however, this is my interpretation rather than the official agency's.
Quality Management System Failures
Software on its own cannot commit fraud or falsification, so enter stage right the intrepid users and their management. These are the main culprits, either through bad practices or deliberate falsification. However, it should be noted that the design of some CDSs makes it easier for people to commit falsification when the data files reside in operating system directories rather than controlled via an integrated database. This should be read in conjunction with an earlier "Questions of Quality" column from April 2012 (10).
It has become clear to inspectors that management and senior management can be responsible for instigating falsification by a variety of means such as direct pressure on analysts to pass material regardless of the results, or lax control of an organization. The overall quality management system (QMS) has been cited in warning letters involving data integrity in the laboratory (12,13). Unlike EU GMP (14), there is no direct US GMP reference to a QMS (8); however, the FDA have taken ICH Q10 on pharmaceutical quality systems (15) and published it as industry guidance (16). Mention of the QMS raises the question of the roles and responsibilities of senior and laboratory management to prevent and detect data falsification. These people set the expectation for their staff to follow — if data falsification is found, management are responsible and often may be culpable. In the Ranbaxy consent decree (17) and in some recent warning letters (12,13,30), FDA advise hiring a third party data integrity consultant. One of their tasks is then to identify the managers who were responsible for falsification; these people will then be disbarred by the agency.
Moreover, the failure of internal audits to identify the problem was highlighted in one warning letter (12). As noted earlier, MRHA requires companies to perform self-inspections under EU GMP Chapter 9 (6) to focus on data integrity within their own organizations and their suppliers (5).
Equipment Citations
A frequent citation in the CDS warning letters is §211.68(b) in the section on automatic, mechanical, and electronic equipment (9), which requires that:
- Access is restricted to authorized individuals.
- Changes are only instituted by authorized individuals.
- The accuracy of calculations must be verified.
- Backups must be exact and complete.
- Backups must be secure from alteration, erasure, or loss.
Non-compliances in this area involve:
- Sharing of user identities between two or more users, thereby making it impossible to identify the individual who was responsible for a particular action within the CDS. It should therefore be ensured that there are enough user licences for each user to have one for their role. Sharing user accounts may seem to be a smart way to save money but, if you get caught, the cost of rectifying the non-compliance makes the saving pale into insignificance. A list of current and historical users is essential for compliance with both Part 11 (18) and Annex 11 (19).
- Access privileges must be appropriate to a user's job function; therefore not everyone can be a system administrator. There will need to be three or more user roles (for example, administrator, user, and trainee) with corresponding access privileges that need to be documented outside of the system either in a configuration specification or a standard operating procedure (SOP).
- The system administrator ideally needs to be independent of the function of the laboratory so that configuration settings cannot be altered and the audit trail turned on and off to hide falsification activities. The system administration activities such as configuration of the software, including controlled changes to it and user account management and access privileges, need to use IT rather than laboratory staff.
- Limit access to both the CDS application software and the workstation operating system as there are many citations for deletion of data in some file-based CDSs.
- Failure to backup data, incomplete backup of data, not having the CDS software to interpret the data files, or just being incompetent and losing data when upgrading the CDS software are just some of the ways companies have been cited under this section of the regulations. The simplest way to avoid this is to give the job of backup to the IT professionals. There are a few catches here: Are the IT staff trained? Is there a backup SOP with evidence of actions? Is recovery tested regularly? Has the backup process and software been validated? This is fine for a networked CDS but if there are standalone workstations then data may be located on the local workstation drive. This is not acceptable and, in my view, a CDS must be networked to avoid the backup problem.
The majority of citations above are where laboratories have standalone workstations.
Citations for Lack of Laboratory Controls
Human inventiveness knows no bounds when it comes to data falsification. One company (20) actually removed some of their chromatographs and workstations from the site to hide data manipulation from inspectors. Other CDS non-compliance citations include:
- Unofficial testing — which we discuss in more detail in the next section.
- Failing to document work contemporaneously. One way this can be achieved is by waiting until the chromatography has been performed, and then working out the sample weight required and falsifying the data (12).
- Overwriting data is possible with some file-based systems and this was used by a number of companies that used older and less compliant CDS applications to hide trial injections to see if a batch would pass or not. In newer CDS, configurable settings can often be set to prohibit the over-writing of data. However when these are not properly established, over-writing of data can occur. Therefore the configuration setting need to be tested in the validation to ensure that this does not occur.
One of the corrective actions requested by the FDA was the writing of an SOP describing a comprehensive computer life cycle to ensure that data integrity was better in the computer systems used by the organization (12).
Failure to Have Complete Laboratory Records
Here's where compliance failures become very interesting. Audit trails in some CDSs were found to be turned off, which is a poor approach to compliance in a regulated environment (12,13,20,21). It is imperative that the audit trail is turned on otherwise changes made to data cannot be attributed to the individual who made it and the old and new values are not recorded. In my view, designers of CDS audit trails must embed them in the basic operation of the system so that they cannot be turned off. The only issue is if the laboratory wants to turn on the reason for change.
When the audit trail in the system was turned on, nobody reviewed the entries (except the inspectors) (1,30) but the audit trail is part of the complete data (9,22,23) that the second reviewer must check.
Other non-compliance citations, as outlined in Figure 1, are reintegration to pass and not saving the integration method. In this case there needs to be technical controls in the CDS software to prevent reintegration without saving the method. In addition, a laboratory needs to have an SOP coupled with training about when it is permissible to reintegrate chromatograms and when it is not.
A common theme with many of the warning letters was the use of trial or test injections (12,13,20,21,30), or unofficial testing. This involves a test injection of samples to check if a batch is going to pass or not. Often the test injections are either conveniently forgotten or, worse, deleted from the CDS as if the test never occurred. This failure to document test injections sits under 211.194(a) (9) for not providing complete data for the analysis (22,23) or raw data (10). This example pertains to the deletion of 5301 data files from a data system (30).
In a citation for Wockardt in November 2013 (21) for using test injections there is the following statement:
"Neither the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) document Q2R, "Validation of Analytical Procedure: Text and Methodology," nor the United States Pharmacopoeia General Chapter <1058>, "Analytical Instrument Qualification," includes instructions for performing "trial" injections for a method that is validated."
This is an interesting citation and rationale. Are these citations appropriate, or just fluff? Let us examine these two references in more detail.
- ICH Q2(R1) (24) outlines the experiments for validation of an analytical procedure. In section 9 there is a single paragraph that outlines the use of system suitability tests (SSTs) for checking that the whole analytical system is suitable for conducting an analysis and cross references the pharmacopoeias for more information.
- United States Pharmacopoeia general chapter <1058> (25) is focused on analytical instrument qualification (AIQ). It is not surprising that it does not mention an operational detail about the test injections because it is not within the scope of the general chapter! Therefore the citation of this reference as justification for not permitting "test" injections is plainly wrong and spurious.
In my view the agency would be on more solid ground if they cited USP <621> on chromatography (26), or even 211.160(a) (9) for scientific soundness.
Is The System Ready to Run?
Let us now look at the issue of "test" injections from another perspective. Do we want to commit samples for analysis when a chromatographic system is not equilibrated? No should be the answer; we want to have a chromatography system ready especially for complex separations or where we analyze at or near the limits of detection or quantification. We therefore have a choice: Do we commit samples for analysis, and if the SST samples fail resolve the problem and start again; or do we have an independent solution to evaluate if a system is ready for the analysis from the outset? Clearly the first option is not optimal and can be a waste of time, especially if the results are required for batch release. However, it can keep the regulators off your back as failing SST results mean that any results generated are not out of specification (OOS) by definition (27).
Let us explore the evaluation injection(s) in a little detail. I'm going to be very clear here, I am NOT advocating injecting aliquots from the vials for the samples under test — this is the quickest way to receive a warning letter. I would argue that under scientific soundness in 211.160(a) (9) the approach for evaluating if a chromatographic system needs a number of criteria can be outlined as follows:
- All chromatographic systems need to equilibrate before they are ready for analysis. The time taken will typically depend on factors such as the complexity of the analysis, the age and condition of the column, and detector lamp warm-up time. Generally there will be an idea of how long this will be from the method development/validation/verification/transfer work performed in the laboratory and this should be documented in the analytical procedure.
- Prepare an independent reference solution of analyte(s) that will be used for the sole purpose of system evaluation. The solution container label needs to be documented to GMP standards and clearly identified for the explicit purpose of evaluating if a chromatography system is ready for a specific analysis.
- The analytical procedure needs to allow the use of system evaluation injections. Staff need to be trained in the procedure.
- Inject one aliquot from the evaluation solution and compare with the SST criteria. Clearly label the vial in the sequence file as a system evaluation injection. If the SST criteria are met then the system is ready for the analysis.
- Upon completion of the analysis, document the number of system evaluation injections as part of the analytical report for the run.
If readers have any alternative approaches that they would like to discuss please send them to me as this would make for a lively debate.
10 CDS Compliance Commandments
Although the focus of this column has been on the role that chromatography data systems have played in cases where fraud and falsification have been discovered by regulatory agencies, it would be remiss of me if I did not use this opportunity to present the way that these systems should be used and the controls that need to be in place to ensure data integrity of the electronic records generated by them and interpreted by chromatographers are trustworthy and reliable.
Therefore, based on this review of warning letters and non-compliances, I have drawn up the 10 CDS compliance requirements and present them in Table 1. As these are relatively self-explanatory I will not discuss them any further in the text.

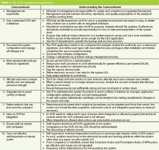
Table 1: Ten CDS compliance requirements.
Summary
This column has focused on the role that chromatography data systems have had in the cases of falsification and fraud that have been discovered by regulatory agencies. The details have been revealed in the warning letters from the FDA and the non-compliances from European regulatory agencies. To ensure that the data and records generated by these systems are trustworthy and reliable, the column concludes with 10 CDS compliance commandments.


Postscript
This column is a prelude to a three-part article looking at what features a CDS should incorporate when used in a regulated environment. It will also look at system architecture, basic functions, and regulatory compliance features.
Acknowledgements
I would like to thank Jennie and Kay McDowall for searching the EU non-compliances and FDA warning letters pages to find the information used in this column.
"Questions of Quality" editor Bob McDowall is Principal at McDowall Consulting, Bromley, Kent, UK. He is also a member of LCGC Europe's editorial advisory board. Direct correspondence about this column should be addressed to the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com
References
(1) Able Laboratories FDA Form 483 Observations, July 2005.
(2) R.D.McDowall, Quality Assurance Journal 10, 15–20 (2006).
(3) Able Laboratories staff criminal prosecution for fraud: http://www.fda.gov/ICECI/CriminalInvestigations/ucm258236.htm
(4) Compliance Programme Guide (CPG) 7346.832 Pre-Approval Inspections, published May 2010, effective May 2012.
(5) MHRA Self Inspections for data integrity: http://www.mhra.gov.uk/Howweregulate/Medicines/Inspectionandstandards/GoodManufacturingPractice/News/CON355490
(6) EU GMP Chapter 9 Self-Inspections (2001).
(7) FDA warning letters: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/default.htm
(8) EMA non-compliances website: http://eudragmdp.ema.europa.eu/inspections/gmpc/searchGMPNonCompliance.do
(9) Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR 211 (2008).
(10) R.D. McDowall, LCGC Europe 25 (4), 194–200 (2012).
(11) R.D. McDowall, LCGC Europe 24 (4), 208–217 (2011).
(12) USV FDA warning letter, February 2014.
(13) Wockardt FDA warning letter, July 2013.
(14) EU GMP Chapter 1 Pharmaceutical Quality Systems (2013).
(15) International Conference on Harmonization (ICH) Q10 Pharmaceutical Quality Systems (2008).
(16) FDA Guidance for Industry, Pharmaceutical Quality Systems (2008).
(17) Ranbaxy Consent Decree of Permanent Injunction, United States Court in the District of Maryland, USA, JFM12CN0250, January 2012.
(18) Electronic records; electronic signatures final rule, 21 CFR 11 (1997).
(19) EU GMP Annex 11, Computerized Systems (2011).
(20) Fresenius Kabi FDA warning letter, July 2013.
(21) Wockardt, FDA warning letter, November 2013.
(22) R.D. McDowall, LCGC Europe 26 (6), 338–343 (2013).
(23) R.D. McDowall, LCGC Europe 26 (7), 389–392 (2013).
(24) International Conference on Harmonization (ICH) Q2(R1) Validation of Analytical Procedures: Text and Methodology (2005).
(25) United States Pharmacopoeia <1058> Analytical Instrument Qualification.
(26) United States Pharmacopoeia <621> Chromatography.
(27) FDA Guidance for Industry Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production (2006).
(28) J. Donath and R.D. McDowall, LCGC Europe 18 (9), 453–464 (2005).
(29) R.D. McDowall, Validation of Chromatography Data Systems: Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, UK, 2005).
(30) Sun Pharmaceutical Industries Limited, FDA Warning letter, May 2014.
(31) http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm







What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






