Rapid Simultaneous Determination of Five Major Alkaloids from Menispermi Rhizoma in Rat Urine by Ultrahigh-Pressure Liquid Chromatography– Tandem Mass Spectrometry (UHPLC–MS/MS) and its Application to a Urinary Excretion Study
Alkaloids are the main bioactive ingredients derived from menispermi rhizoma (MR), which is often used in traditional Chinese medicine. However, simultaneous determination of different types of alkaloids in urine has not been reported. A sensitive, rapid, and efficient ultrahigh-pressure liquid chromatography–tandem mass spectrometry (UHPLC–MS/MS) method for rapid quantification of five major bioactive alkaloids (covering three different types) in rat urine has been established. Extraction of five alkaloids from rat urine was performed with acetonitrile precipitation. The separation was carried out on a C18 column, and the detection was performed with positive electrospray ionization (ESI) MS in multiple reaction monitoring (MRM) mode. The method was fully validated and successfully applied to a urinary excretion study of five alkaloids in rats after they were given an oral dose of MR extract. It is the first time that the urinary excretion profiles of dauricoside, acutumine, and acutumidine in rats has been reported. The results obtained lay the foundation for the clinical application and safety evaluation of MR.
Menispermi rhizoma (MR) is the dried rhizome of Menispermum dauricum DC, which is a well-known Chinese herb and widely used in traditional Chinese medicine. It is reported in the Chinese Pharmacopoeia that the herb can treat sore throats, enteritis dysentery, and rheumatism arthralgia (1). In the last few decades, much attention has been focused on the chemical composition and pharmacological studies of MR. According to the literature, the ethanol extract of MR exhibits a variety of pharmacological effects, such as anti-inflammatory (2,3), anti-bacterial (4), antioxidant (5), anti-hypoxia (6), anti- tumor (7), anti-arrhythmia (8) effects, and the protection of injury induced by myocardial-cerebral ischemia (9,10). These pharmacological activities are presumed to result from the presence of various bioactive ingredients in MR.
Some phytochemical research on MR showed that it contains a large number of alkaloids, which include bisbenzylisoquinoline, protoberberine, and morphinane (11). Among these alkaloids, dauricine, dauricicoline, dauricoside, acutumine, and acutumidine (shown in Figure 1) are not only the most abundant alkaloids in MR, but also the most physiologically active ones. The biological activities of the five alkaloids have been widely reported. Dauricine has shown obvious effects on inhibiting blood platelet aggregation (12) and neuroprotection (13). Dauricine has also shown effects on anti-arrhythmia and anti-tumors (14). Moreover, according to the Chinese Pharmacopoeia, dauricine is used as the only component to assess the quality of MR. The antiarrhythmic effect of dauricine has been verified in several animal models, and it is widely used in cardiac arrhythmia patients (15). Meanwhile, dauricine exhibited a significant reverse use-dependent effect by blocking the Na+, K+, and Ca2+ ion currents of cardiac transmembranes (16). It is reported that dauricine can suppress hypoxia-inducible factor 1-alpha (HIF-1a) protein accumulation and vascular endothelial growth factor (VEGF) expression to inhibit cancer angiogenesis. Our previous study demonstrated that dauricicoline had a protective effect on hypoxia injury of EAhy 926 cells (6). Dauricoside has been proven to possess significant selective affinity to dopamine receptor D1 (17) and an inhibitory effect on blood platelet aggregation caused by adenosine 5’-diphosphate (12). Both acutumine and acutumidine belong to morphinanes, which have multiple biological activities. For example, they have significant antihypoxia effects (6). Acutumine has been reported to possess selective cell cytotoxity (18). Acutumidine can also inhibit the production of hepatitis B surface antigen (HBsAg), and the IC50 value is 2.023 mM (19). To date, a large number of research studies on the pharmacology of MR have been carried out.


FIGURE 1: The chemical structures of the five active alkaloids and the IS.
Generally, synergy among multiple constituents in vivo plays an important role in the pharmacological action of traditional Chinese medicines (20). Thus, simultaneous determination of multiple bioactive ingredients in vivo is crucial to illustrate the pharmacological and clinical effects of MR. Studies on MR in vivo still remain scarce; studies have mainly focused on the determination of these alkaloids in plasma (21–23). Urine excretion of drugs is essential to investigation of interpreting in vivo dispersion, which is also closely related to the efficacy and safety of drugs (24). To date, only two articles have reported on the excretion of dauricine (25,26). As a result, there is no successful approach for simultaneous determination of the five alkaloids in urine because of the lack of reference standards. Meanwhile, a urinary excretion study of the five alkaloids after oral administration of MR extract has not been reported. This is a great obstacle to further investigation of safe administration of MR in clinical applications. Thus, it is urgent to establish an available bioanalytical method for simultaneous determination of the five alkaloids in urine samples.
Currently, several analytical approaches, such as capillary electrophoresis (CE) (27), high-performance liquid chromatography (HPLC) with UV detection (28), diode array detection (DAD) (29), and tandem mass spectrometry (MS/MS) (30) have been employed to quantify alkaloids in MR. Among these methods, HPLC-UV, HPLC– MS/MS, and UHPLC–MS/MS have been used to quantify alkaloids from MR in rat plasma (21–23,25). UHPLC–MS/MS combined with multiple reaction monitoring (MRM) mode has attracted much attention and become increasingly popular in bioanalysis because of its unique advantages, such as high speed, high selectivity, and strong specificity. Thus, the purpose of this study was to develop a rapid and sensitive UHPLC–MS/MS approach in MRM mode for simultaneous determination of five alkaloids (covering three different types) in rat urine and to apply it in a urinary excretion study. It is the first report on a UHPLC–MS/MS assay of five alkaloids in urine after dosing of MR extract. The results could be useful for the further studies on the usage, dosage, mechanisms of action, and pathology and toxicology of MR.
Materials and Methods
Reagents and Materials
The reference standard of dauricine was obtained from Shenzhen Meihe Biological Technology Co., Ltd. In our laboratory, dauricicoline, dauricoside, acutumine, and acutumidine were obtained from MR, and their structures were identified by physicochemical data and spectroscopic analysis (MS, 1H, and 13C NMR spectra) (6). The purity of the reference standards were all above 97.0%, as determined using area normalization by HPLC. The MR was purchased from Anguo (Hebei, China) and authenticated by Professor Jincai Lu (School of Chinese Materia Medica, Shenyang Pharmaceutical University). A voucher specimen (Number 20141001) has been deposited in the Department of Pharmaceutical Analysis at Shenyang Pharmaceutical University. The internal standard (IS), verapamil hydrochloride with a purity of 99.00%, was obtained from the National Institutes for Drug and Food Control (batch number: 100223-200102, Beijing, China). HPLC grade acetonitrile and methanol were supplied by Fisher Scientific. Formic acid of HPLC grade was also obtained from Sigma-Aldrich. Tetrahydrofuran was purchased from Concord Technologies. Ultrapure water was purchased from Wahaha Corporation Ltd.
UHPLC–MS/MS Conditions
The liquid chromatography–triple quadrupole mass spectrometer consisted of an Acquity UPLC system and a Xevo TQ-S API triple quadruple mass spectrometer system (Waters Corporation, Milford, Massachusetts, USA). The chromatographic separation of analytes and IS was conducted on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm, 1.7 μm). A Vanguard BEH C18 column (5 mm × 2.1 mm, 1.7 μm) acted as the guard column. The column temperature was kept at 35 °C. Gradient elution employed a mobile phase of 0.1% formic acid in water (A) and acetonitrile (B) with a flow rate at 0.3 mL/min. The gradient elution was programmed as follows: 0–2.0 min, 8% B; 2.0–4.0 min, 8–20% B; 4.0–8.0 min, 20–60% B; and 8.0–10.0 min, 60–8% B. The injection volume was 5 μL.
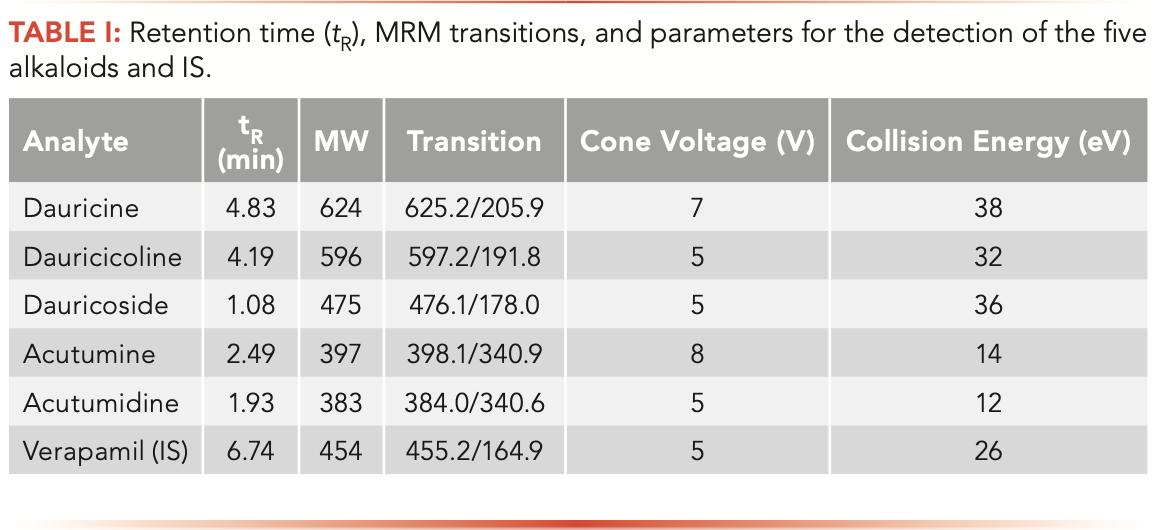
Analyte detection was conducted using positive ion electrospray ionization (ESI) in the MRM mode. Nitrogen was used as both nebulizing and drying gas. The optimized instrument parameters were as follows: The capillary voltage was set at 3.0 kV; the source temperature was 150 °C; the desolvation temperature was 350 °C; and the desolvation and cone gas flow were set at 700 and 150 L/h, respectively. The cone voltage, collision energy, and MRM transitions for each analyte and IS varied; their specific parameters were optimized and are displayed in Table I. Chromatograms and data acquisition were performed using a data qualitative and quantitative analysis system.

Preparation of MR Extract
MR powder (500 g) was mixed with 5.0 L ethanol:water (95:5, v/v) and refluxed twice (1.0 h each time). Then, extraction with ethanol:water (75:25, v/v) was performed twice using the same method. The extraction solutions were filtered and concentrated under vacuum. To calculate the quantity of drug administered, the amounts of five alkaloids in MR extract were measured by the external standard method. The contents of dauricine, dauricicoline, dauricoside, acutumine, and acutumidine were 23.19, 3.63, 0.711, 0.167, and 0.762 mg/g, respectively.
Preparation of Standards and Control Solutions
Stock solutions of dauricine, dauricicoline, dauricoside, acutumine, and acutumidine were separately prepared by dissolving the compounds in tetrahydrofuran and then precisely diluted with methanol to achieve final concentrations of 0.1 mg/mL. Then, an appropriate amount of the five stock solutions was mixed and gradiently diluted with methanol to obtain a linear concentration of standard working solutions. The working solution of the IS at 10.0 ng/mL was obtained from the stock solution.
Calibration standards and quality control samples were prepared in a glass tube with tapered bottom by evaporating 100 μL working solutions to dryness with nitrogen (N2) and then mixing with a 100 μL of blank urine. The ranges of urine concentration were 5 to 2500 ng/mL for dauricine; 2.4 to 1200 ng/mL for daurici- coline; 2 to 1000 ng/mL for dauricoside; 6 to 3000 ng/mL for acutumine; and 10 to 5000 ng/mL for acutumidine, respectively. Quality control (QC) samples were prepared at three concentration levels of 10, 141, and 2000 ng/mL for dauricine; 4.8, 68, and 960 ng/mL for dauricicoline; 4, 57, and 800 ng/ mL for dauricoside; 12, 170, and 2400 ng/mL foracutumine; and 20, 283, and 4000 ng/mL for acutumidine.
Urine Sample Preparation
A simple protein precipitation (PPT) method was used for the extraction of five alkaloids from rat urine. First, 20 μL of IS solution was put into glass tubes and evaporated to dryness with N2. Next, 100 μL of the urine sample was transferred to the glass tube and vortexed for 1 min. Then, 500 μL of acetonitrile was added. After vortexing for 2 min, the mixture was centrifuged for 10 min at 15,000 rpm. The supernatant was removed to another polyethylene tube and evaporated to dryness with N2. The residues were reconstituted with 100 μL of 50% aqueous acetonitrile. After vortexing for 2 min and centrifuged for 10 min at 15,000 rpm, a 5 μL aliquot of supernatant was injected for UHPLC–MS/MS analysis.
Method Validation
The method was validated according to the guidelines set by the United States Food and Drug Administration (FDA) for bioanalytical method validation (31).
Selectivity was evaluated by analyzing the chromatograms of six individual blank rat urine samples, blank urine samples spiked with the five analytes and IS, and the actual urine samples obtained from the excretion study after administration of MR extract.
Calibration samples were prepared and assayed in duplicate over three consecutive days. The calibration curve of seven concentration levels was constructed by plotting the peak area ratios (y) of each analyte to IS versus urine sample concentrations (x) of the analytes, using a weighted 1/x2 linear regression. The limit of detection (LOD), depicted as the concentration of analyte, gave a signal-to-noise (S/N) ratio of three. The lower limit of quantification (LLOQ), depicted as the concentration of analyte giving a S/N ratio above 10, was set at the lowest concentration of the calibration curve. The accuracy of the LLOQ was within ±20% and the relative standard deviation (RSD) was within 20% (n = 6).
Six replicates of quality control (QC) samples at three concentration levels were used to evaluate the intra- and inter- day (three days) precision and accuracy of the method. The precision at each concentration level was expressed as relative standard deviation (RSD) of the measured concentration and accuracy as the relative error (RE) of the measured mean value deviating from the nominal value. According to the FDA guidance, the RSD determined at each concentration level must not exceed 15% and RE within ±15%.
The recovery of five alkaloids and IS was measured by comparing the peak responses of five alkaloids and IS from extracted QC samples with those of samples spiked in post-extracted blank urine at an equivalent concentration. The matrix effects of rat urine for five alkaloids and IS were evaluated by comparing the peak response of post-extraction spiked samples with those from the non-extracted neat standard solutions at equivalent concentration. Both recovery and matrix effects of the five analytes were determined at three concentration levels, whereas for the internal standard, the recovery and matrix effect were determined at a single concentration of 2.0 ng/mL by determining six replicates of QC samples.
The stability of the five alkaloids in rat urine was determined by analyzing QC samples in six replicates at three concentration levels. The short-term stability of the analytes in rat urine was determined after keeping the spiked samples at room temperature for 8 h. Long-term stability was assessed after keeping the QC samples at −80 °C for two weeks. Freeze–thaw stability in rat urine was investigated after three freeze–thaw cycles. The post-preparation stability was evaluated after keeping the extracted QC samples in an auto-sampler at 10 °C for 12 h. The stability of the standard solutions (analytes and IS) was assessed by comparing the determined concentration of the solutions kept at 25 °C for 4 h and 12 h, and 4 °C for one month with that of the freshly prepared solutions, respectively.
When the concentrations of urine samples are higher than the upper limit of quantitation (ULOQ) of each analyte, it is required to dilute them with blank urine. To validate the dilution process, a 10x dilution of urine samples were assessed in six duplicates. The diluted QC samples were prepared at a concentration above the ULOQ sample level and diluted 10-fold with blank rat urine.
To confirm that carryover had no effect on the accuracy and precision of the assay, carryover effect was evaluated by six consecutive injections of a blank sample after the injection of an ULOQ sample.
Excretion Study
The described UHPLC–MS/MS method was applied to determine the urine concentrations of the 5 alkaloids after dosing of MR extract. Six male Wistar rats (220–260 g) were supplied by the Animal Center of Shenyang Pharmaceutical University. The rats had free access to food and water and were acclimatized in an environmentally controlled breeding room before experimentation. Under the above conditions, all the rats were acclimated for at least one week and then fasted for 12 h prior to experimentation. All experiments were done in strict accordance with the Regulations of Experimental Animal Administration. The MR extract was dissolved in 0.5% carboxymethyl cellulose sodium (CMC-Na) solution and was orally administered at a dose of 5.77 g/kg (equivalent to 133.78 mg/kg of dauricine, 20.95 mg/kg of dauricicoline, 4.10 mg/kg of dauricoside, 0.96 mg/kg of acutumine, and 4.40 mg/kg of acutumidine). Afterward, the rats were individually put in metabolic cages to collect rat urine. Urine samples were collected before administration and at the time intervals of 0–4, 4–8, 8–12, 12–24, 24–48, 48–72, 72–96, 96–120, and 120–144 h post dosing. The urine volume collected in each interval was recorded. All sample supernatants collected were stored at −80 °C until further analysis.
Results and Discussion
Optimization of the UHPLC–MS/MS Conditions
In this study, a BEH C18 column (50 mm × 2.1 mm, 1.7 μm, Waters) compatible with low liquid flow rate was selected, which offered several attractive benefits, including higher peak intensity, lower solvent consumption, and a shorter runtime. Using this column, methanol and acetonitrile organic phases were investigated. A higher mass spectrometry (MS) response and lower background noise was obtained with mobile phase of acetonitrile. Water containing different proportions of formic acid, acetic acid, and ammonium formate was tested to improve peak shapes of analytes. Finally, water containing 0.1% formic acid was adopted and higher sensitivity in positive ESI for the alkaloids was achieved.
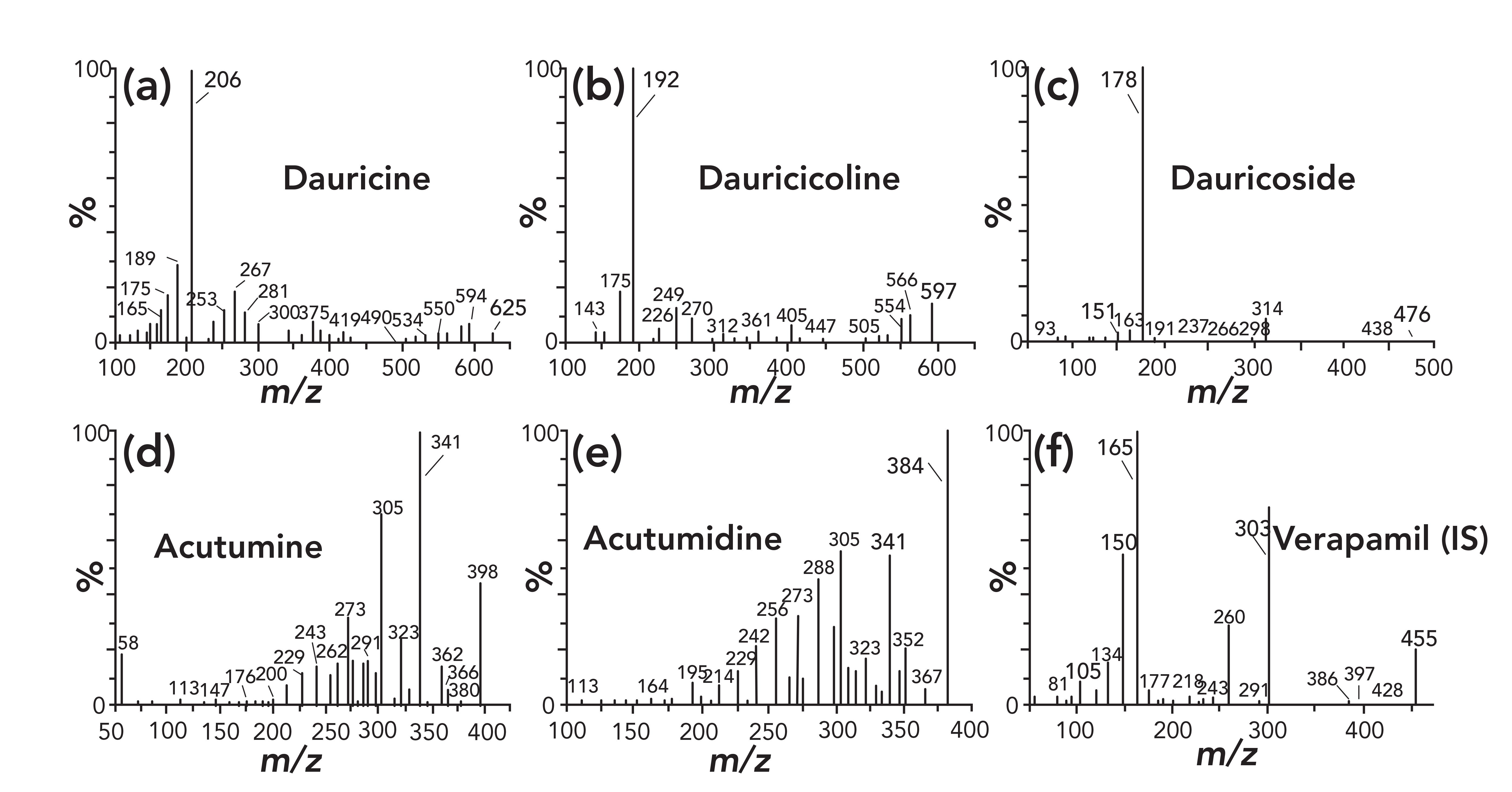
To achieve optimum ionization efficiency and sensitivity of each analyte and the IS, the MS parameters were optimized using the standard solutions. Given that all the analytes and IS contain nitrogen, the full-scan mass spectra revealed a higher S/N ratio for both the analytes and IS in positive mode. Stable molecular ion peaks for dauricine, dauricicoline, dauricoside, acutumine, acutumidine, and IS were obtained at m/z 625.2, 597.2, 476.1, 398.1, 384.0 and 455.2, respectively. Then collision energy and cone voltage were optimized to obtain the corresponding product ion spectra for each analyte. The MS/MS spectra of the five analytes and IS are shown in Figure 2. The MS fragmentation behavior of analytes was very similar because they have the same structural skeleton. Dauricine and dauricicoline have the abundant product ion at m/z 206 and 192 from their molecular ion peaks. For acutumine and acutumidine, the m/z 340 product ion was more abundant and selected for MRM transition, while the optimum product ion of dauricoside was m/z 178 (Table I).

FIGURE 2: Product ion mass spectra of 5 active components and the IS in positive electrospray ionization mode: (a) dauricine; (b) dauricicoline; (c) dauricoside; (d) acutumine; (e) acutumidine; (f) verapamil (IS). Axes labels for all figures are m/z for x-axis and percent (%) for y-axis.
Sample Preparation Optimization
Sample processing plays an important role in bioanalytical assays. To improve recovery and minimize endogenous interference, both PPT and liquid–liquid extraction (LLE) were investigated in this study. The results indicated that low extraction recovery (especially for dauricoside) was obtained when rat urine was processed by LLE. Thus, the PPT method was adopted. Methanol and acetonitrile were tested as precipitation reagents. Eventually, acetonitrile proved to be a better extraction solvent for the five analytes.
Method Validation
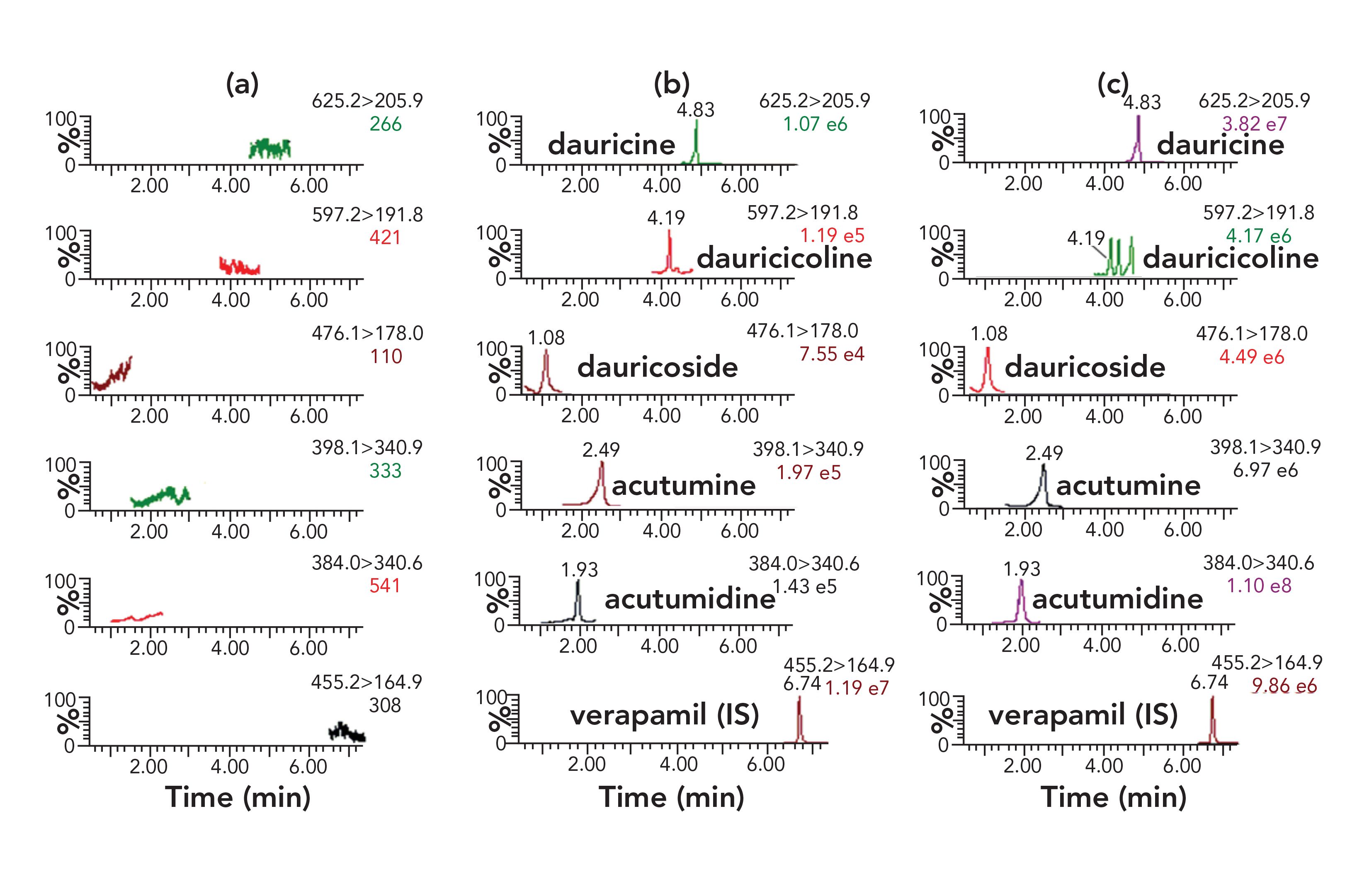
Representative MRM chromatograms of blank urine, blank urine spiked with the target compounds (at LLOQs) and IS, and a urine sample after dosing are shown in Figure 3. No interfering peaks were detected at the retention times of five analytes and IS.

FIGURE 3: Typical MRM chromatograms of the 5 active components and IS in rat urine: (a) blank rat urine sample; (b) blank rat urine spiked with the 5 analytes at LLOQ and IS; (c) rat urine 0-4 h samples collected after oral administration of menispermi rhizoma extract (5.77 g/kg). Axes labels for all figures are time (min) for x-axis and percent (%) for y-axis.
The standard calibration curves, linear ranges, linearity (r) and LLOQs of the 5 alkaloids are listed in Table II. The calibration curves showed good linearity with a correlation coefficient r ≥ 0.9902. The LODs for dauricine, dauricicoline, dauricoside, acutumine, and acutumidine were 1.5, 0.6, 0.6, 1.8, and 3.0 ng/mL, respectively. The LLOQs for dauricine, dauricicoline, dauricoside, acutumine, and acutumidine were 5.0, 2.4, 2.0, 6.0, and 10.0 ng/mL, respectively, which is sensitive enough to explore the excretion behaviors of the analytes.

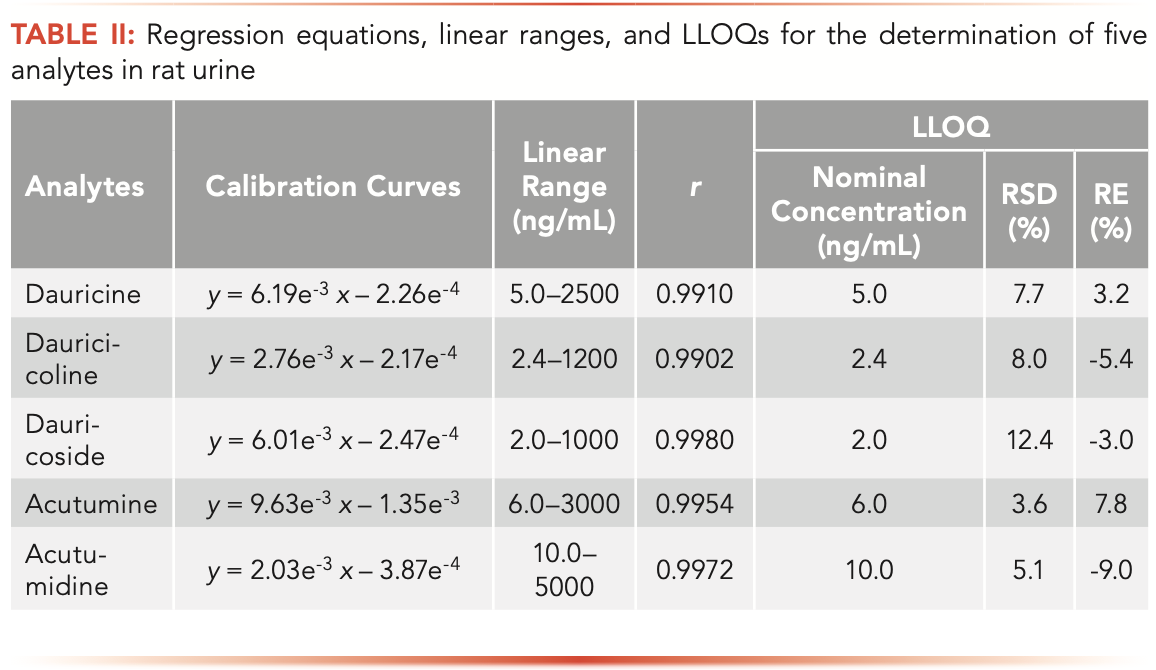
The summary results for precision and accuracy of five alkaloids are listed in Table III. The intra-day precision varied from 2.6% to 12.4%, while the inter-day precision varied from 2.1% to 12.0%. The intra- and inter-day accuracy ranged between -10.2% and 11.2%. It was found that both precision and accuracy values were all within the acceptable criteria (±15%, except ±20% at LLOQ).

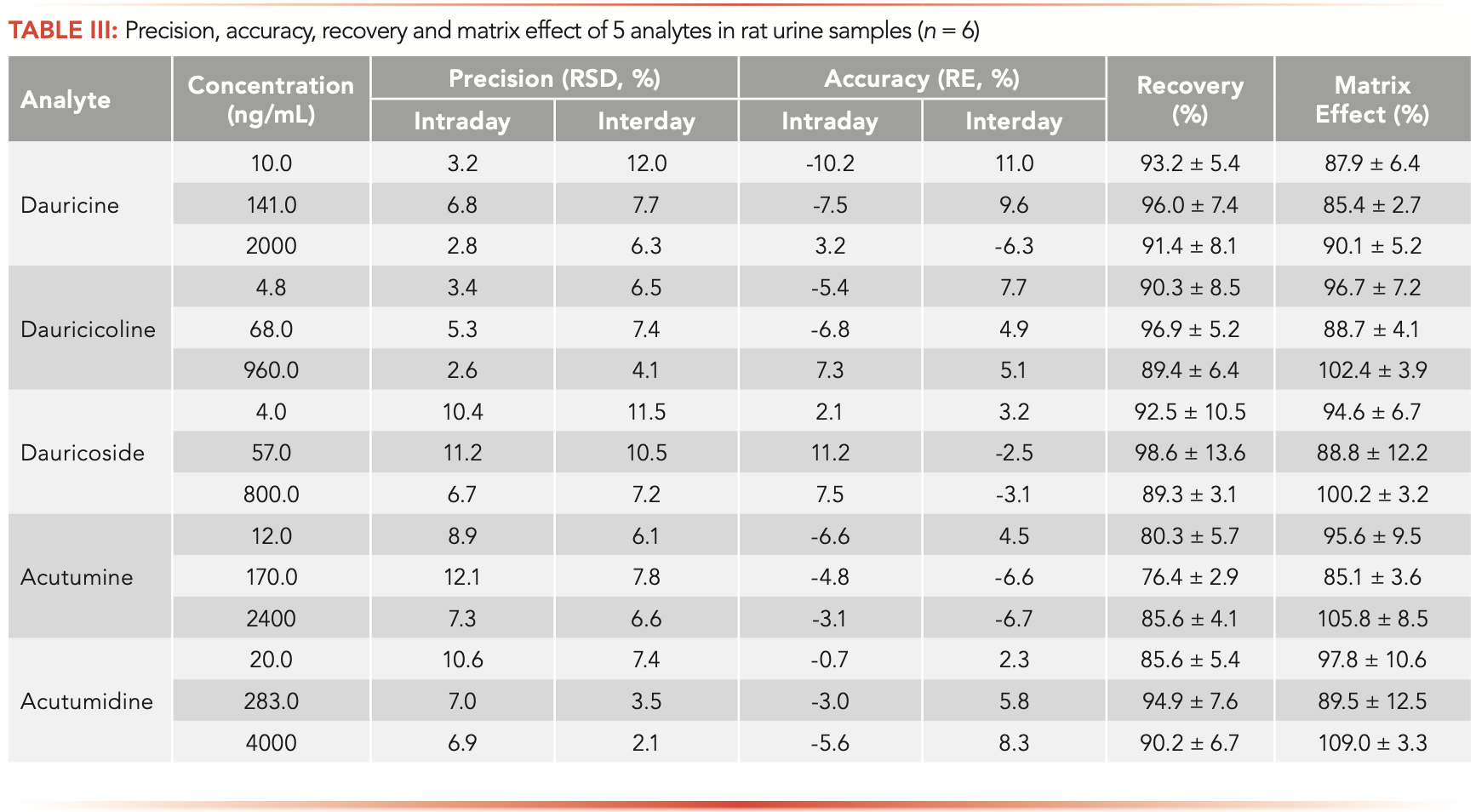
The extraction recoveries and matrix effects of the five analytes and IS in rat urine are listed in Table III. The mean recoveries of dauricine, dauricicoline, dauricoside, acutumine, and acutumidine from rat urine were 91.4–96.0%, 89.4–96.9%, 89.3–98.6%, 76.4–85.6%, and 85.6–94.9% at the three QC levels. The mean recovery of IS normalized verapamil was 87.4% at a concentration of 2.0 ng/mL. The mean matrix effects of rat urine for analytes and IS ranged from 85.1–109.0%. Both the extraction recoveries and matrix effects met the acceptance criteria of the FDA.
The results of the standard solutions stability study indicated that the standard solutions of the analytes were stable for at least one month at 4 °C (RE: −4.3% to 3.5%, RSD <4.5%). The stability results of the analytes in rat urine are summarized in Table IV. It was found that the analytes were stable under various processing conditions including 10 °C for 12 h, three freeze-thaw cycles, 8 h at room temperature, and −80 °C for two weeks.

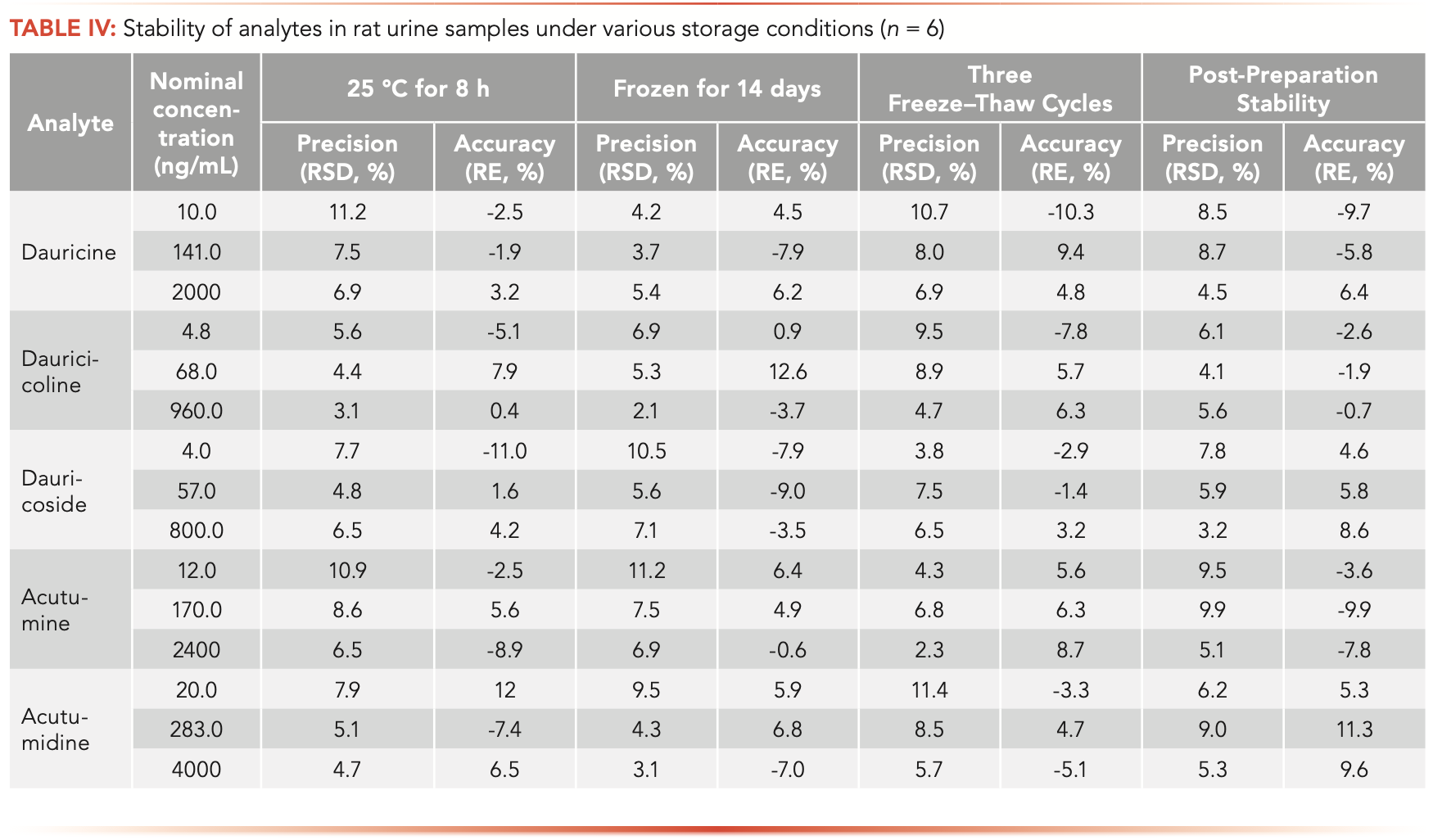
The precision and accuracy of the dilution integrity were validated. The results showed that precision and accuracy at each concentration were within the acceptable criteria (RSD <15%; RE ±15%). The samples with concentrations greater than the ULOQ could be met by appropriate dilution with blank matrix.
When injecting blank sample after ULOQ samples, no signal at the retention time of the analytes and IS was detected. The results indicated that the carryover from residues was negligible.
Urinary Excretion Study
The newly established UHPLC–MS/MS approach was applied to the rat urinary excretion study of the five alkaloids after dosing of MR extract (5.77 g/kg). The mean cumulative excretion-time profiles of the five alkaloids in rat urine are displayed in Figure 4. Results showed that five alkaloids were mainly excreted within 144 h after oral dosage. The cumulative excretion amount of the five alkaloids in urine over the 144 h period were 335118 ± 63965 ng for dauricine, 54564 ± 13915 ng for dauricicoline, 8860 ± 2019 ng for dauricoside, 16371 ± 5155 ng for acutumine, and 83130 ± 24784 ng for acutumidine.

FIGURE 4: Mean cumulative urinary excretion-time profile of 5 analytes in rats after oral administration of menispermi rhizoma extract (n = 6). Axes labels for all figures are time (min) for the x-axis and cumulative excretion (ng) for the y-axis.
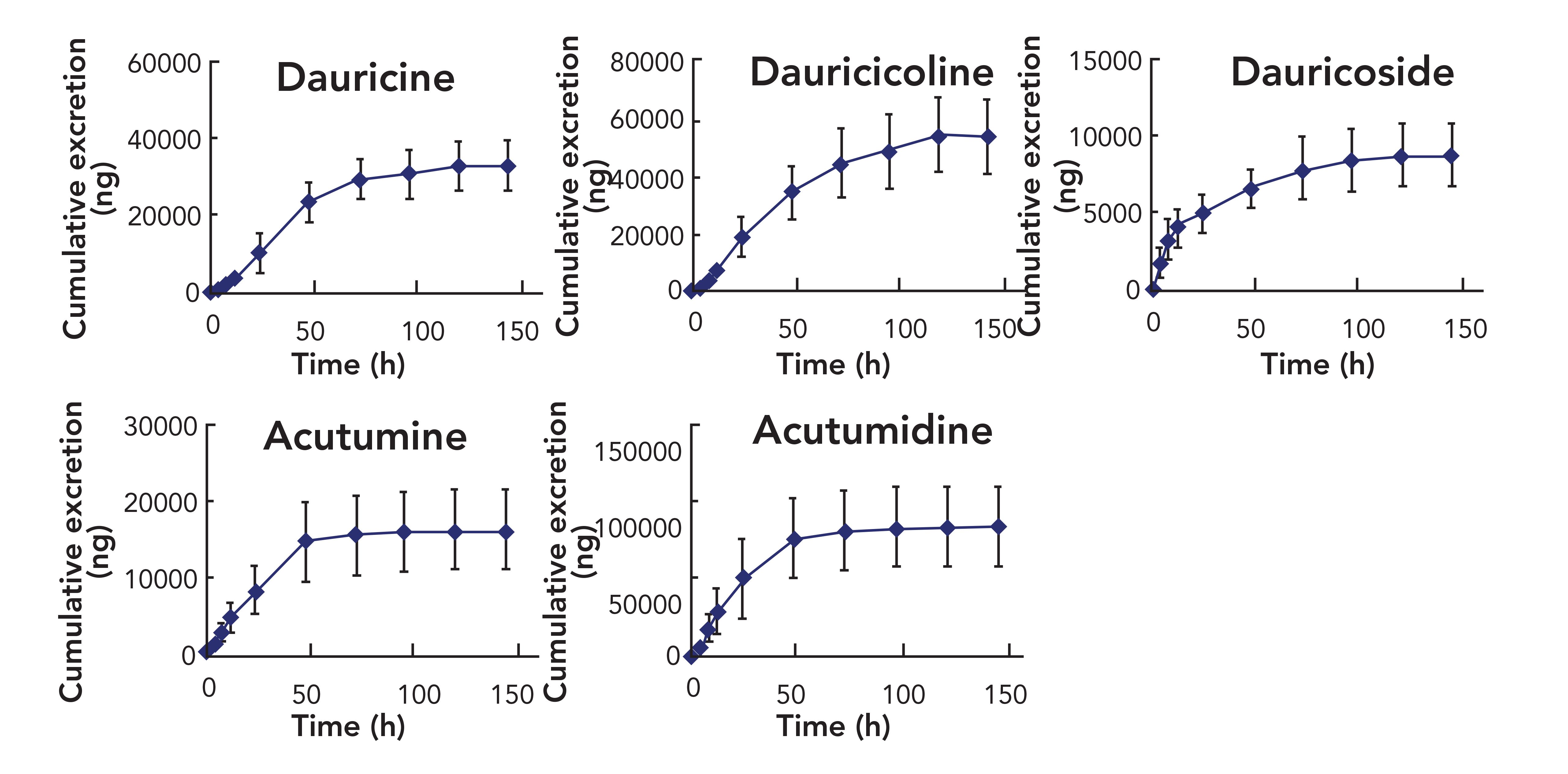
Dauricine and dauricicoline exhibited a similar urinary excretion characteristic because they have the same structural skeleton. There was significant increase for them in their urine excretion after 12 h, and the rate of excretion continued to increase from 12 h to 48 h. The cumulative excretion of the two analytes reached to a plateau at 72 h after oral administration. Compared with the dosage, the recoveries for dauricine and dauricicoline within 144 h were about 1.255% and 1.302%, respectively. The results suggest that dauricine and dauricicoline were mainly eliminated as metabolites in urine. In addition, the urine excretion characteristic of dauricine has been reported previously (25); the results of the report in the literature were higher than the results of this study. It is speculated that the other components in MR may promote the metabolism of dauricine in vivo. In terms of dauricoside, there was a significant increase in urine excretion from 0 h to 4 h, and the cumulative excretion of dauricoside reached a plateau at 72 h after oral administration.
The cumulative urine excretion amount accounted for 1.080% of the total dose. Perhaps it was extensively metabolized under the action of intestinal enzymes and the excretion forms of dauricoside were its metabolites (including its aglycone). Thus, further experiments on its metabolism will need to be conducted. Both acutumine and acutumidine are morphine alkaloids, and showed a similar urinary excretion process. The excretion rate increased significantly from 12 to 24 h after dosing, and reached a plateau at 48 h. Total recoveries of acutumine and acutumidine over the 144 h period were about 8.50% and 9.45% of the total dose, respectively. Compared with the above three alkaloids, the morphine alkaloids have a higher excretion ratio in the prototype form, which may be attributed to their higher polarity. In this study, the urine excretion characteristics of dauricoside, acutumine, and acutumidine were reported for the first time.
Conclusion
This newly established UHPLC–MS/MS bioanalytical method was rapid, efficient, and highly sensitive, with a short run time of 10.0 min, and met all the requirements in bioanalysis of the FDA bioanalysis guideline. The analytical method was successfully applied to excretion studies of the five alkaloids following administration of MR extract. This is the first time to report of excretion studies of dauricine, dauricicoline, dauricoside, acutumine, and acutumidine together in vivo after administration of MR extract. We believe that the described results will be of value in accelerating further research on the mechanism of action of MR and will provide useful information for the clinical application and development of MR.
Acknowledgments
This research was supported by National Natural Science Foundation of China (Grant No. 81703690) and Natural Science Foundation of Tianjin (No 19JCQNJC12200).
References
(1) Chinese Pharmacopoeia Commission, Chinese Pharmacopeia (China Medical Science Press, Beijing Part I), 99 (2015).
(2) Q. Su, J. He, Z.Y. Wang, L. Lv, Y. Suo, J.J. Wang, Z.W. Zheng, C.C. Huo, and J. Li, J. Ethnopharmacol. 193, 12–20 (2016).
(3) D. Sun, M.G. Zhou, X.H. Ying, B.F. Cheng, Y.Q. Han, Y. Nie, Y.Y. Hou, and H.G. Bai, BMC Complem. Altern. Med. 14, 356 (2014).
(4) J. Li, H.Y. Bi, B.T. Du, Y.Z. Sun, and H.L. Zhang, J. Qufu Normal Univ. 36, 96–98 (2010).
(5) T. Yi, Y.J. Jin, J.J. Jia, and X.F. Li, J. Yanbian Univ. 43, 128–130 (2017).
(6) J. Shao, C.F. Shi, J.X. Wei, Y.X. Li, and X.J. Guo, China J. Chin. Mater. Med. 44, 723–729 (2019).
(7) D. Wu, J. Du, Y. Zhang, Y. Sun, and H. Zhang, J. Cancer Res. Ther. 14, S505– 511 (2018).
(8) X.H. Liu and F.L. Han, Heilongjiang Med. J. 13, 160–162 (2000).
(9) X.J. Zhang, L.J. Guo, L. Qu, and Q. Lv, Acta Pharm. Sin. 39, 661–665 (2004).
(10) B. Zhao, Y. Chen, X. Sun, M. Zhou, J. Ding, J.J. Zhan, and L.J. Guo, Molecules 17, 2725–2737 (2012).
(11) X.Q. Zhang, W.C. Ye, S.X. Zhao, and C.T. Che, Phytochemistry 65, 929–932 (2004).
(12) S.M. Hu, S.X. Xu, X.S. Yao, C.B. Cui, Y. Tezuka, and T. Kikuchi, Chem. Pharm. Bull. 41, 1866–1868 (1993).
(13) Z. Wang, J. Wang, Y. Guo, T. Lu, X. Wang, and Q. Duan, Neural Regener. Res. 4, 15–19 (2009).
(14) J. Wang, Y. Li, X.B. Zu, M.F. Chen, and L. Qi, Asian Pac. J. Trop. Med. 5, 973–976 (2012).
(15) J.S. Xia, Z. Li, J.W. Dong, H. Tu, and F.D. Zeng, Acta Pharmacol. Sin. 23, 371–375 (2002).
(16) J.Q. Qian, Acta Pharmacol. Sin. 23, 1086–1092 (2002).
(17) B.W. Yu, J.Y. Chen, Y. He, G.Z. Jin, and G.W. Qin, Chin. J. Nat. Med. 9, 249–252 (2011).
(18) B.W. Yu, J.Y. Chen, Y.P. Wang, K.F. Cheng, X.Y. Li, and G.W. Qin, Phytochemistry 61, 439–442 (2002).
(19) P. Cheng, Y.B. Ma, S.Y. Yao, Q. Zhang, E.J. Wang, M.H. Yan, X.M. Zhang, F.X. Zhang, and J.J. Chen, Bioorg. Med. Chem. Lett. 17, 5316–5320 (2007).
(20) R.J. Liu, L. Zheng, M.L. Cheng, Y. Wu, P. Gu, Y.J. Liu, P.C. Ma, and L. Ding, J. Chromatogr. B 1014, 83–89 (2016).
(21) J.X. Wei, L.L. Fang, X.L. Liang, D. Su, and X.J. Guo, Talanta 144, 662–670 (2015).
(22) Y.X. Jin, C.C. Song, L.J. Shao, L.T. Wang, X.T. Tu, B.B. Chen, J.Y. Chen, Y.H. Zhi, C.C. Wen, and W.Z. Zhu, Lat. Am. J. Pharm. 36, 1204–1209 (2017).
(23) X.Y. Liu, Q. Liu, D.M. Wang, X.Y. Wang, P. Zhang, H.Y. Xu, H. Zhao, and H.Q. Zhao, J. Chromatogr. B 878, 1199–1203 (2010).
(24) Y. Xie, G. Zhong, H. He, G. Fan, and Y. Wu, J. Pharm. Biomed. Anal. 54, 148–153 (2014).
(25) S.J. Chen, Y.M. Yang, Y.M. Liu, B. Zhang, X.B. Pang, and F.D. Zeng, Chin. Pharmacol. Bull. 17, 225–229 (2001).
(26) J. Yu, B.L. Zhu, D. Su, and Z. Jiang, RSC Adv. 8, 31633–31645 (2018).
(27) J.M. Wang, H.Y. Zhai, Z.G. Chen, Q.W. Li, and J.J. Mo, Chin. J. Pharm. Anal. 25, 1329–1332 (2005).
(28) D. Su, Z.K. Wang, T.T. Sun, and X.J. Guo, J. Shenyang Pharm. Univ. 1, 46–50 (2016).
(29) Y.N. Liu, X. Song, R.Q. Yan, T.X. Li, X. Chai, A.D. Qi, Y.F. Wang, and Z.Z. Jiang, J. Food Drug Anal. 21, 206–218 (2013).
(30) J.X. Wei, K.N. Cui, Y.Y. Du, J. Yu, Z. Jiang, and X.J. Guo, Anal. Methods 9, 3029–3038 (2017).
(31) U.S. Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation (2001).
Jinxia Wei, Yanan Li, and Yubo Li are with the School of Chinese Materia Medica at the Tianjin University of Traditional Chinese Medicine, in Tianjin, China. Yingying Yu is with the Department of Health Service at the Logistics College of Chinese People’ s Armed Police Forces, in Tianjin, China. Xingjie Guo is with the School of Pharmacy at the Shenyang Pharmaceutical University, in Shenyang, China. Direct correspondence to: yuboli1@163.com






 and its Application to a Urinary Excretion Study")












Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

