Radical Mass Spectrometry as a New Frontier for Bioanalysis
LCGC Europe
In this article, we discuss radical MS, an increasingly important area of MS development and the application to bioanalysis. At the current stage, most of the research is performed by a small set of academic groups; it is likely that these types of fundamental studies will attract more attention and even be commercialized in the near future.
In this instalment, we discuss radical ion chemistry as an increasingly important area of mass spectrometry (MS) development and the application to bioanalysis. At the current stage, most of the research is performed by a small set of academic groups. Given the unique capability offered by radical MS compared to the traditional studies on even-electron ions of analytes, it is likely that these types of fundamental studies will attract more attention and even be commercialized in the near future.
Radicals are atoms, molecules, or ions that contain one or more unpaired valence electrons or an open electron shell. They are, generally, highly reactive because of their need to convert themselves to more stable, even-electron species. Well-controlled radical chemistry can provide the means for conducting some of the most difficult and delicate chemical transformations, like converting RNA to DNA by ribonucleotide reductase, an enzyme with a radical as its catalysis centre. Radical chemistry can be coupled with mass spectrometry (MS) to tackle traditionally challenging problems such as sequencing disulphide proteins and determining C=C location in lipids. Here, we discuss this increasingly important area of MS development and its application to bioanalysis.
Background
MS has been established as a powerful tool for qualitative and quantitative chemical analysis. Its principle of analysis involves at least three crucial steps for extracting as much information as possible from an analyte: Forming ions from the analyte molecule of interest, performing mass-to-charge ratio (m/z) analysis (so-called mass analysis), and perturbing ions to induce m/z changes. It typically relies on tandem mass spectrometry (MS–MS), a process in which ions are allowed to interact with neutral molecules, electrons, ions, electromagnetic waves, and so on.
During the past several decades, these three steps have reflected significant advances. The development of various soft-ionization methods permits the transformation of almost any molecule or chemical entity from its original physical state to gas-phase ions with the least change to its structure. (Nevertheless, it is frequently necessary to add a charge carrier to the analyte, facilitating MS analysis.)
Several types of mass analyzers, the component of mass spectrometers that separate ions according to their mass-to-charge ratios, have been developed and commercialized. Instruments with time-of-flight (TOF) analyzers offer a fast speed of analysis. Fourier transform ion-cyclotron resonance (FT-ICR) and orbital ion trap instruments offer high-performance mass measurement and achieve a resolution in millions and sub-parts-per-million mass accuracy. Accelerator MS (AMS) instruments offer superb dynamic range (108 –1015) for isotope analysis. Finally, some instruments could even be made portable, miniaturized for performing field analyses.
For tandem MS, many activation and dissociation options are available, including collision-induced dissociation (CID) and electron-based excitation or dissociation methods. Clearly, MS has entered into a golden age of advanced development and can be readily applied to solving complicated problems, whether in the areas of drug discovery, cancer research, environmental monitoring, or exploring Mars.
Some of our most pressing questions about MS concern whether its usefulness has been fully exploited and, if not, what its next major advance will be. Its history shows that innovation in instrumentation and inquiries in new gas-phase ion chemistry often went side-by-side, providing the impetus for additional MS development. We believe the scope of gas-phase ion chemistry is unlimited, that its exploration so far has been only marginal, and that it may therefore give rise to the next major MS development.
An area of increasing attention is the development of gas-phase, radical-ion chemistry for bioanalysis. Radical ions, which consist of unpaired electrons, derived from biomolecules (peptides, proteins, lipids, and carbohydrates) are significantly less investigated than even-electron ions. The available ionization methods do not lend themselves to directly forming the radical ions of biomolecules, so additional reaction steps are required to effect the transformation from even-electron ions. Several research groups have invested a major effort in developing methods to produce radical ions of biomolecules. Given the coexistence of two reactive functional groups within one ion, a charge and a radical site, the chemistry of radical ions is rich. It differs significantly from the chemistry of even-electron ions. This difference can be exploited to provide structural information that complements the structural information gleaned from even-electron ions. A well-known example is the development of electron-capture dissociation (ECD) and electron-transfer dissociation (ETD). These techniques provide rich structural information for protein analysis that complements the information from CID. Thus, it substantially improves the capability of protein identification and characterization.
Bio-radicals have been implicated as important intermediates in a wide variety of biochemical processes. At the molecular level, they are associated with oxidative damage to proteins, DNAs, and lipids, which have been shown to be related to ageing, neurodegenerative diseases, and cancers. Several classes of enzymes also use radicals for catalysis. For example, ribonucleotide reductase uses a free radical (thiyl radical) for its catalytic activity. Interestingly, the radical storage centre is tens of amino acid residues away from the catalytic centre. It is not clear, at this point, how the enzyme tightly regulates radical reactivity along this long-range radical transfer process. In general, knowledge of the bio-radical species is constrained by the limited techniques available to analyze these reactive intermediates. Studying the gas-phase chemistry of bio-radicals, therefore, provides direct experimental evidence of their intrinsic chemical properties (reactivity, energetics, and structure), which can help explain the fate, including intra molecular and intermolecular radical transfer, of bio-radical species after they initially form.
The following discussions provide examples of methods of forming bio-radical ions of different classes of biomolecules and how radical chemistry and MS can generate useful structural information.
Peptides and Proteins
Table 1 summarizes the major types of amino acid or peptide radical ions that have been studied, together with their methods of formation. They are categorized as follows, according to their underlying mechanisms of radical-ion formation:
- Electronic state change of even-electron species via electron capture, electron transfer, electron detachment, interactions with high-energy photons, or collisions with meta-stable neutrals.
- Intramolecular electron transfer in peptide-metal-ligand complexes.
- Homolytic cleavage of labile bonds (either by CID or photolysis).
- Radical reactions of disulphide-linked peptides in the plume of electrospray ionization (ESI).


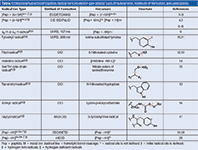
Table 1: Representative list of peptide radical ions studied in gas phase; type of radical ions, methods of formation, and precursors.
When an electron adds to an even-electron peptide cation (multiply protonated), it results in more hydrogen than the corresponding even-electron species. Subsequent dissociation of the hydrogen-rich radical ions produces N-Cα backbone fragments, as compared with amide-bond cleavages of even-electron ions from vibrational activation (21–23). Analytical advantages of fragmenting hydrogen-rich radical ions include less dependence on peptide sequences and preservation of the labile modification on amino acid residues. ECD (1,24) and ETD (2,19) are the two major methods using fragmentation of the hydrogen-rich radical ions and have been shown to be effective for protein analysis by providing structural information complementary to that available by the CID of even-electron ions.
Fragmentation of hydrogen-deficient radical ions, which (when compared with even-electron peptide ions) evidence fewer hydrogen atoms, has also been studied. Their dissociation chemistry differs from that of hydrogen-rich species and is sensitive to the nature of radicals (25). Several research groups have contributed significantly to this area by developing unique methods of forming hydrogen-deficient radical ions and studying their gas-phase ion chemistry (Table 1). It has been shown that radical-dissociation chemistry can be analytically useful in protein analysis by inducing site-specific fragmentation (26,27) differentiating structural isomers or stereoisomers (17,28), and probing labile post-translational modifications (PTMs) (29).
An alternative approach to generating the peptide radical ions used by our group involves a radical addition to a peptide ion, to form peptide radical ions, as shown in equation 1. Except for proton-transfer reactions, other possible reaction routes, such as electron-transfer and functional-group abstraction by radical species, could lead to the formation of peptide radical ions:

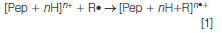
This approach also gives rise to the formation of hydrogen-deficient radical ions, which facilitates choosing the proper radical-reaction chemistry for target peptides, thus conferring the flexibility to produce different types of radicals (such as carbon-centred versus heteroatom centred radicals) with highly specific radical sites. Moreover, because the charging process of the peptide ions can be separated from the radical formation step, this approach makes it possible to manipulate the charge properties of radical ions — the charge states, charge carriers (proton or metal ions), and charge polarities.
The capability of forming site-specific radicals is essential for studying radical migration in biomolecule systems, whereas manipulation of the charge property is useful in investigating its role in peptide radical chemistry.
Our group recently applied radical reactions in the nanoelectrospray ionization (nanoESI) plume region for the formation of sulphur-centred peptide radical ions. The experimental setup involves a radical-inducing source: an atmospheric-pressure, low-temperature, helium-plasma source or a low-pressure Hg lamp in the proximity of a nanoESI plume (Figure 1). A high number density of OH radicals can be generated and subsequently reacted with disulphide-linked peptides entrained in the spray plume. Given the high reactivity of the disulphide bond towards radical species, cleavage at the bond is highly favourable. This preference results in the well-controlled formation of sulphur-centered radicals such as the cysteine sulphinyl radical (-SO•), thiyl radical (-S•), and perthiyl radical (-SS•).

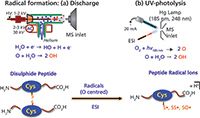
Figure 1: Radical reactions in the nanoESI plume involving disulphide-linked peptides to form sulphur-centred peptide radical ions.
These peptide radical ions can be mass isolated in a mass spectrometer for further investigation. Note that these three types of radical species have also been detected in enzymatic systems, though little chemistry is known because of their transient nature in condense phases. With their formation in the gas phase, the intrinsic chemical properties lend themselves to extensive study.
Ion and molecule reactions in a quadrupole ion trap show that the reactivity of the three types of sulphur radicals follows the order of thiyl (RS•) > perthiyl (RSS•) > sulphinyl (RSO•). Although the cysteine sulphinyl radical is quite inert under conventional bimolecular reaction conditions, collisional activation facilitates intramolecular reactions. These reactions reveal novel chemistry, because the supplied external energy overcomes the energy barrier. Two newly discovered reaction channels involve the exchange of the cysteine sulphinyl radical (SO•Cys), with either a free thiol (-SH) or a disulphide bond (S-S) within a peptide ion system (reactions shown in Figure 2). Note that, in a protein system, the exchange of the sulphinyl radical with a disulphide bond leads to disulphide-bond scrambling. These reaction phenomena are significant, for they may affect how a radical intermediate migrates in a protein system and attacks other functional groups, resulting in conformational or functional changes. In addition, the reactions suggest that the oxidative damage — damage done by forming a sulphinyl radical — can potentially be repaired by reacting with a nearby thiol group (Figure 2[a]).

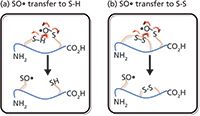
Figure 2: Intramolecular cysteine sulphinyl radical (SO
•Cys) transfer with (a) a free thiol and (b) a disulphide bond.
Disulphide bond formation is a critical PTM, which is highly relevant to the structural integrity of a protein and its biological function. An MS method targeting an intact, disulphide peptide or protein is desirable because it can provide linkage information about disulphide bonds and rich sequence information. It is worth noting that cleavage of the disulphide bond within an even-electron peptide ion is typically a higher-energy process under collisional activation, as compared with cleavage of the peptide backbone (amide bond) to produce protein-sequence ions. To produce a fragment ion from the peptide backbone region within a disulphide bond loop requires the cleaving of at least two bonds. Thus, few sequence fragment ions form under CID conditions, making sequence information difficult to obtain. The routine procedure in proteomics, therefore, involves disulphide reduction and alkylation, to open the disulphide bonds. Indeed, the sequence information obtained from CID-based tandem MS can be significantly improved, but only at the cost of losing information about disulphide-bond connecting patterns.
Unlike the chemistry of even-electron ions, when a radical is introduced into the peptide or protein system, the disulphide bond presents itself as an easy target. For instance, its cleavage is a favourable process where radical intermediates are produced, in ECD, ETD, electron-detached dissociation (EDD), and UV-photodissociation. Among those means of dissociation, ETD is increasingly applied for disulphide peptide or protein characterization. Facile cleavage at the disulphide bond and separation of chains originally connected by disulphide bridges have been reported from ETD of interchain, disulphide-linked peptides. For peptides consisting of intrachain disulphide bonds, ETD can produce rich-sequence information even from backbone regions covered by the disulphide bone loop. This facility of EDT is the result of radical cascades in which N-Cα bond cleavage from ETD leads to the formation of c and z• ions, and the carbon-centred radical z• can further react and cleave the disulphide bone to release c/z, c + 32/z - 32, c -33/z + 33 types of sequence ions, all of which are useful for peptide identification.
Radical reactions in the ESI plume (setup shown in Figure 1) also efficiently cleaves the disulphide bond. One approach to achieving enhanced sequence information for the disulphide peptide combines radical reactions in the ESI plume with subsequent MS2-CID, to fragment peptide radical ions, thus obtaining the traditional b/y type fragment ions (Figure 3). We applied this method to a series of naturally occurring peptides that contained one or multiple disulphide bonds. In doing so, we obtained significantly enhanced sequence information, as compared with CID of the intact disulphide peptide ions. Using insulin, a polypeptide containing three disulphide bonds, as an example, rich-sequence information corresponding to 75% of backbone cleavages is obtained from radical reactions in ESI and subsequent CID, which compares favourably to the reported ECD and ETD results (~60%). Moreover, this result is significantly more favourable than the 26% backbone cleavages obtained from CID of intact, disulphide, peptide ions.

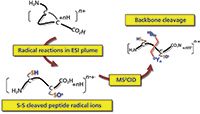
Figure 3: Coupling radical reactions in the ESI plume region with a subsequent MSâMS scan of the disulphide-bonded, cleaved-peptide, radical ions to improve sequence information.
Lipids
Lipids serve in numerous biological functions such as structural binding blocks, energy storage, and signalling transduction. The degree of a lipid's unsaturation (the number of C=C bonds) as well as the positions of the C=C bonds determine its overall structure and therefore its biochemical and biophysical properties. For instance, n-3 polyunsaturated fatty acids (PUFAs) (also called omega-3 fatty acids, in which 3 is the double-bond position counted from n, the terminal methyl group) are essential for the functional development of the brain and retina. By contrast, no such effects have been observed for n-6 PUFAs. The efforts to determine double-bond positions in lipids, however, are not trivial, largely because of a need to distinguish the correct structure from a large number of possible double-bond-position isomers. In the field of lipidomics, therefore, MS is the analytical method of choice, because of its high sensitivity, its selectivity, and its ability to provide detailed structural information. Tandem MS of even-electron lipid ions based on collisional activation is useful in providing the head group and acyl-chain composition information. Yet because cleavages at the C-C and C=C positions require significantly higher energies than other possible fragmentation channels, such as neutral or charged head-group losses and fragmentation at the ester bond, obtaining C=C position information cannot be achieved directly from low-energy collisional activation. When the collision energy is high enough (that is, ~ keV from sector instruments or a TOF-TOF instrument), charge-remote fragmentation is possible. Thus, the C-C bonds adjacent to the C=C bonds are cleaved, so the location of C=C is inferable. Compared with the brute-force method of simply increasing the level of collision energy, reactions targeting the unique chemistry of C=C have been used, including ozonolysis, methoxylation, and olefin cross-metathesis. When coupled with MS analysis (on-line or off-line), these chemistry types provide information about the location of C=C.
Another unique property of C=C is its susceptibility to radical attack, which prompts investigation by radical-involved MS analysis. For example, Blanksby and colleagues explored radical-directed dissociation (RDD) of lipids by attaching a photo-caged radical precursor to the target analyte (30). They found that irradiation of the precursor in the mass spectrometer releases the previously caged radical, and CID of the nascent radical and lipid complex ion yields fragmentation information. Moreover, analyzing the fragment ions and fragmentation patterns can lead to ascertaining the number of unsaturation and the position of double bonds.
Our group recently explored coupling a Paternò–Büchi (P-B) reaction of C=C in lipids in the ESI plume with subsequent tandem MS, for fast and sensitive determination of the C=C location (31). The P-B reaction is a classic [2+2] photochemical reaction. In organic synthesis, it is widely used to form compounds containing an oxetane ring. The reaction mechanism involves UV excitation of the carbonyl group within an aldehyde or ketone to a ketyl diradical, which subsequently reacts with the C=C in an olefin. Depending on the relative positions of the carbonyl and the C=C bond, two position isomers of the oxtanes can be formed, as shown in Figure 4. When energy (such as heat) is applied to the P-B reaction products, retro P-B reactions proceed through two possible pathways. One route leads to the original reactants. The other leads to cleavage of the C-C bond at the initial C=C bond position and the C-O bond of the initial carbonyl group, forming a new olefin and ketone or aldehyde. The latter pathway is of special interest in this study because the reaction products carry the C=C bond's position information.

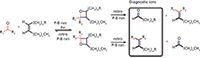
Figure 4: PaternòâBüchi (P-B) reaction between ketone, aldehyde, and olefin in lipids together with retro P-B reactions, to form diagnostic ions for the determination of C=C location.
The P-B reaction can be simply executed in the nanoESI plume region by means of the setup shown in Figure 1, a UV lamp with the irradiation wavelength centred at 254 nm. The lipid analyte is dissolved in 1:1 (water:acetone), where acetone functions as the P-B reaction reagent. A reasonable reaction yield of 40% is possible for different classes of lipids, and no reaction selectivity towards specific C=C locations or configurations is found. The P-B reaction product is stable and can be mass isolated and subjected to CID. Figure 5 shows an example of the P-B reaction of oleic acid (C18:1 9Z) prepared at 10 µM in 1:1 water–acetone, with 1% ammonium hydroxide, for nanoESI. Abundant P-B reaction product (m/z 339.3), because of the addition of acetone to the C=C, was observed upon UV irradiation. MS2-CID of the P-B reaction product promotes the retro P-B reactions and provides a pair of "diagnostic ions" (m/z 171 and 197) with a mass difference of 26 Da, for confident determination of the C=C bond location. The structures of the diagnostic ions are shown in Figure 5(d). The method is sensitive (~femtomolar [fmol] for fatty acid) and can be readily coupled with "shotgun" lipidomics, for complex lipid-mixture analysis. An in-solution P-B reaction has been developed to couple with on-line liquid chromatographic separation methods.

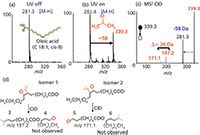
Figure 5: PaternòâBüchi (P-B) reaction between oleic acid and acetone in the nanoESI plume upon UV irradiation. MS1 spectra of oleic acid (a) before UV irradiation, (b) upon UV irradiation, in which the P-B reaction product is located at m/z 339.3, and (c) MS2-CID of the P-B reaction product. The diagnostic ions (m/z 171.1 and 197.2) for the C=C location are observed. (d) Structures of the diagnostics ions (structures 3 and 5). The structures explain the origin of the 26-Da mass difference.
In general, the P-B reaction method for lipid C=C location determination has analytical advantages. They include simple experimental setup for reactions, no need to modify the mass spectrometer, easy-to-interpret mass spectra, and inexpensive derivatizing reagents. These characteristics should make this method accessible and attractive to many laboratories.
Oligosaccharides
Saccharides are highly susceptible to decomposition by free-radical processes. In consideration of this fact, Beauchamp and colleagues (32) proposed using biomimetic synthetic reagent and free-radical chemistry to analyze saccharides. By introducing a radical at the reducing end of the saccharides, both glycosidic bond cleavage and cross-ring cleavages are observed upon CID of radical-precursor ions. Radical-driven fragmentation pathways are proposed as responsible for fragments arising from both types of cleavages. This method for the analysis of saccharides, based on free radicals, also represents a novel method for understanding the reactions between free radicals and saccharides.
Summary and Future Directions
Radical-involved chemical and biochemical reactions are attracting increased interest in the area of MS. This interest is driven by the distinct chemistry of radicals versus even-electron species, and the potential for new analytic capabilities. This chemistry can be successfully applied for the structural analysis of proteins or peptides, lipids, and saccharides. The extent of structural information obtained from MS analysis of these molecules can be greatly improved relative to conventional methods. The development of radical MS approaches and strategies has only just started. We anticipate its continued growth, both in fundamental ion chemistry and method and instrument development. We are confident that radical MS can serve as one of the new forces advancing the future development of MS in chemical analysis.
Xiaoxiao Ma, PhD, earned his bachelor's degree in environmental engineering in 2007 from Tsinghua University in Beijing, China. In 2012, advised by Xinrong Zhang, he earned a PhD in analytical chemistry from the same institution. As a visiting PhD candidate from 2009–2010, he spent the year in Professor Yu Xia's lab working on gas-phase ion/radical reactions involving disulphide peptides. Ma is currently a post-doctoral fellow under Professors Xia and Zheng Ouyang (Purdue Biomedical Engineering, West Lafayette, Indiana, USA). He is working on lipid-structure analysis by means of mass spectrometry.
Yu Xia, PhD, earned her bachelor's degree in chemistry from Lanzhou University, in China, in 1999. In 2002, she earned a master's degree in organic chemistry from Shanghai Institute of Material Medica, Chinese Academy of Sciences and in 2006 she earned a PhD in analytical chemistry from Purdue University, West Lafayette, Indiana, USA, under the supervision of Scott A. McLuckey. R. Graham Cooks guided her post-doctoral training, and she is currently an assistant professor of chemistry at Purdue. Her research focuses on developing new mass spectrometry methods and tools based on radical ion chemistry. Xia was also the recipient of the 2013 ASMS Research Award.
Kate Yu, "MS — The Practical Art" Editor, joined Waters in Milford, Massachusetts, USA, in 1998. She has a wealth of experience in applying LC–MS technologies to various application fields such as metabolite identification, metabolomics, quantitative bioanalysis, natural products, and environmental applications. Direct correspondence about this column should be addressed to the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com
References
(1) R.A. Zubarev, N.L. Kelleher, and F.W. McLafferty, J. Am. Chem. Soc. 120, 3265–3266 (1998).
(2) J.E. Syka, J.J. Coon, M.J. Schroeder, J. Shabanowitz, and D.F. Hunt, Proc. Natl. Acad. Sci. U.S.A. 101(26), 9528–33 (2004).
(3) S.L. Cook, O.L. Collin, and G.P. Jackson, J. Mass Spectrom. 44(8), 1211–1223 (2009).
(4) I.K. Chu, C.F. Rodriguez, F. Rodriguez, A.C. Hopkinson, and K.W.M. Siu, J. Am. Soc. Mass Spectrom. 12(10), 1114–1119 (2001).
(5) I.K. Chu and J. Laskin, Eur. J. Mass Spectrom. 17(6), 543–556 (2011).
(6) B.A. Budnik, Y.O. Tsybin, P. Hakansson, and R.A. Zubarev, J. Mass Spectrom. 37(11), 1141–1144 (2002).
(7) R.A. Zubarev, M.L. Nielsen, and B.A. Budnik, Eur. J. Mass Spectrom. 6(3), 235–240 (2000).
(8) C.L. Kalcic, T.C. Gunaratne, A.D. Jonest, M. Dantus, and G.E. Reid, J. Am. Chem. Soc. 131(3), 940–942 (2009).
(9) L.Y. Zhang and J.P. Reilly, J. Am. Soc. Mass Spectrom. 20(7), 1378–1390 (2009).
(10) Q.Y. Sun, H. Nelson, T. Ly, B.M. Stoltz, and R.R. Julian, J. Proteome Res. 8(2), 958–966 (2009).
(11) T. Ly and R.R. Julian, J. Am. Soc. Mass Spectrom. 20(6), 1148–1158 (2009).
(12) V. Ryzhov, A.K.Y. Lam, and R.A.J. O'Hair, J. Am. Soc. Mass Spectrom. 20(6), 985–995 (2009).
(13) G. Hao and S.S. Gross, J. Am. Soc. Mass Spectrom. 17(12), 1725–1730 (2006).
(14) J. Steill, J.F. Zhao, C.K. Siu, Y.Y. Ke, U.H. Verkerk, J. Oomens, R.C. Dunbar, A.C. Hopkinson, and K.W.M. Siu, Angew. Chem., Int. Ed. 47(50), 9666–9668 (2008).
(15) S. Wee, A. Mortimer, D. Moran, A. Wright, C.K. Barlow, R.A.J. O'Hair, L. Radom, and C.J. Easton, Chem. Commun. 40, 4233–4235 (2006).
(16) D.S. Masterson, H. Yin, A. Chacon, D.L. Hachey, J.L. Norris, and N.A. Porter, J. Am. Chem. Soc. 126(3), 720–721 (2003).
(17) I.K. Chu, J. Zhao, M. Xu, S.O. Siu, A.C. Hopkinson, and K.W.M. Siu, J. Am. Chem. Soc. 130, 7862–7872 (2008).
(18) A. Kalli and K. Hakansson, Int. J. Mass Spectrom. 263(1), 71–81 (2007).
(19) J.J. Coon, J. Shabanowitz, D.F. Hunt, and J.E.P. Syka, J. Am. Soc. Mass Spectrom. 16(6), 880–882 (2005).
(20) H.J. Yoo, N. Wang, S. Zhuang, H. Song, and K. Hakansson, J. Am. Chem. Soc. 133(42), 16790–16793 (2011).
(21) E.A. Syrstad and F. Tureček, J. Am. Soc. Mass Spectrom. 16(2), 208–224 (2005).
(22) F. Tureček and E.A. Syrstad, J. Am. Chem. Soc. 125(11), 3353–3369 (2003).
(23) M. Sobczyk, W. Anusiewicz, J. Berdys-Kochanska, A. Sawicka, P. Skurski, and J. Simons, J. Phys. Chem. A 109(1), 250–258 (2005).
(24) R.A. Zubarev, N.A. Kruger, E.K. Fridrikkson, M.A. Lewis, D.M. Horn, B.A. Carpenter, and F.W. McLafferty, J. Am. Chem. Soc. 121, 2857–2862 (1999).
(25) A.C. Hopkinson, Mass Spectrom. Rev. 28(4), 655–671 (2009).
(26) T. Ly and R.R. Julian, J. Am. Chem. Soc. 130(1), 351–358 (2008).
(27) J.K. Diedrich and R.R. Julian, J. Am. Chem. Soc. 130(37), 12212–12213 (2008).
(28) Y.Q. Tao, N.R. Quebbemann, and R.R. Julian, Anal. Chem. 84(15), 6814–6820 (2012).
(29) J.K. Diedrich and R.R. Julian, Anal. Chem. 83(17), 6818–6826 (2011).
(30) H.T. Pham, T. Ly, A.J. Trevitt, T.W. Mitchell, and S.J. Blanksby, Anal. Chem. 84, 7525–7532 (2012).
(31) X. Ma and Y. Xia, Angew. Chem. 126(10), 2630–2634 (2014).
(32) J. Gao, D.A. Thomas, C.H. Sohn, and J.L. Beauchamp, J. Am. Chem. Soc. 135(29), 10684–10692 (2013).













Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


The Effect of Time and Tide On PFAS Concentrations in Estuaries
April 8th 2025Oliver Jones and Navneet Singh from RMIT University, Melbourne, Australia discuss a recent study they conducted to investigate the relationship between tidal cycles and PFAS concentrations in estuarine systems, and offer practical advice on the sample preparation and LC–MS/MS techniques they used to achieve the best results.



