Analysis of Four Phthalate Monoesters in Human Urine Using Liquid Chromatography Tandem Mass Spectrometry
LCGC Europe
A fast, sensitive and accurate quantitative method was developed using HPLC–MS–MS for analysis of phthalate metabolites in urine samples.
A method was developed using high performance liquid chromatography with tandem mass spectrometry (HPLC–MS–MS) detection for the analysis of phthalate metabolites in urine samples. The urine samples were treated using solid-phase extraction columns. Multiple reaction monitoring models were used for quantitative detection with high sensitivity and selectivity. In this work, a fast, sensitive, and accurate quantitative method is provided to detect toxic phthalate esters in urine and assess environmental toxicants.
Phthalate esters are widely used as plasticizers in manufacturing. Phthalate esters are also used as raw materials of pesticide carriers, paint, adhesives, cosmetics, lubricants, and so on (1). Because phthalate esters are noncovalently bound to the plastic and maintain relatively independent chemical properties, they can leach out of these products and regularly release into the environment as time goes on (2,3). Globally, more than six million tonnes of phthalates are used each year (4). Presently, phthalate esters have been detected in many different areas in the ecological environment of the world's major industrial countries; they have become one of the most popular global pollutants. This has severely threatened the environment and human health, despite the fact that phthalate esters play an important role in the production and life of mankind (5).



Diet, skin, and inhalation are considered the major exposure routes for phthalate esters (6). When phthalate esters enter the human body they are rapidly metabolized and hydrolyzed to the corresponding phthalate monoesters before absorption, which are the biologically active molecules and recognized testicular toxicant (7,8), and then further oxidized into various metabolites (9). General metabolic pathways of phthalates in humans are shown in Figure 1 (10). Most phthalate esters and their metabolites are excreted in urine or faeces. The total concentrations of phthalate metabolites in urine are normally used as biomarkers of individual intake of phthalate esters (11). The monoesters contain a free reactive carboxylic acid that can conjugate with α-D-glucuronic acid to produce more hydrophilic compounds. Phthalate esters, which mimic oestrogen, have been reported to affect multiple biochemical processes as environmental hormones in humans and wildlife. It is one of the major reasons for male reproductive problems (12–14). Excessive exposure to phthalate esters can increase the risk of breast cancer for women. Many phthalate esters can damage the reproductive systems of offspring as well (15–18). For instance, studies showed that some toxicologically active metabolites, such as MEHP, MBPA, and MBzP, are capable of crossing rodent placenta, which causes adverse effects on the development of the male reproductive system and induces early embryonic death, respectively (19,20). Animal experiments have shown that the toxicities of some monoesters are greater than those of corresponding diesters (21).


Phthalate monoesters are considered to be valuable biomarkers for exposure to phthalate esters. Measuring phthalate monoesters investigates the contamination status, absorption, and metabolism of phthalate esters in the body better than phthalate diesters. Several developed methods have been used to quantify phthalate monoesters, including gas chromatography–mass spectrometry (GC–MS) (22,23). Calafat developed a sensitive method for measuring 13 phthalate metabolites in breast milk using isotope dilution high performance liquid chromatography (HPLC) with negative ion electrospray ionization tandem mass spectrometry (24). This method showed good reproducibility and accuracy. Liquid chromatography–tandem mass spectrometry (LC–MS–MS) methods with high selectivity and sensitivity show an advantage compared to GC–MS methods because of simple sample pretreatment without derivatization.

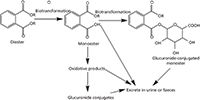
Figure 1: General metabolic pathways of phthalates in humans. R = H; phthalic acid = PA, C2H5; monoethyl phthalate = MEP, (CH2)3CH3; monobutyl phthalate = MBP, CH2C6H5; monobenzyl phthalate = MBzP, CH2CH (C2H5) (CH2)3CH3; mono (2-ethylhexyl) phthalate = MEHP.
Some methods, such as on-line solid-phase extraction (SPE) coupled to HPLC–MS–MS, are considered the most advanced method to investigate phthalate esters and their metabolites, but the high cost limited their widespread application. To develop a sensitive, selective, low cost, and efficient analytical method to quantify these chemicals appears essential for understanding how human health might be affected by exposure to phthalates under general laboratory conditions. In this article, we report a method for the quantitative detection of four phthalate monoesters including MEP, MBP, MBzP, and MEHP simultaneously in human urine samples. The method involves the enzymatic dissociation of conjugated monoesters, SPE, and separation and detection using HPLC with negative ion electrospray ionization tandem mass spectrometry. The method was used to analyze human urine samples collected in the same area for evaluating potential phthalate ester exposure.
Experimental
Materials: PA, MEP, MBP, MBzP, MEHP (>99.9%), and their isotope-labelled internal standards (>99.9%) were purchased from AccuStandard, Inc. Methanol, acetonitrile, and ethyl acetate were purchased from Sigma Aldrich Laboratories, Inc. β-glucuronidase (Escherichia coli-K12) was purchased from Roche Biomedical. Ammonium acetate (1 M, pH 6.5), formic acid solution (0.1 M), acetic acid solution (0.1%, v/v), and phosphate buffer solution (pH 2.0) were prepared. Water was purified using a Milli-Q gradient A10 system (Millipore). SPE columns were purchased from Varian, Inc. (Nexus ABS Elut, 60 mg/3 mL). Human urine samples (pooled from 84 individuals) were collected in one area and stored at -80 °C. In this study, plastic equipment was not used in any sampling or experimental processes to avoid contamination, and all glass apparatus were washed with chromic acid solution and rinsed with deionized water and methanol before drying.
HPLC Conditions: Chromatographic separations were performed on a Waters Quattro Micro LC–MS–MS system (Waters Corporation) with a 150 mm × 2.1 mm Kromasil 100-5C18 column (Akzo Nobel) set at 40 °C. Mobile-phase A was 0.1% acetic acid in water, and mobile-phase B was 0.1% acetic in acetonitrile. The mobile-phase gradient is shown in Table 1. The sample injection volume was 20 µL. The flow rate was 0.3 mL/min.

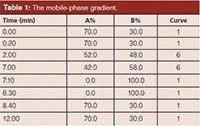
Table 1: The mobile-phase gradient.
Mass Spectrometer Conditions: The MS detector was used in the negative electrospray ionization (ESI) mode. The optimum operation conditions were as follows: ionization voltage: -2.85 kV; cone voltage: 20 V; ion source temperature: 100 °C; drying gas temperature: 300 °C; electron multiplier voltage: 650 V; and collision energy: 16–19 eV. A multiple reaction monitoring (MRM) model was used. The detection channels are shown in Table 2.


Table 2: Linearity of phthalate ester analytes.
Sample Preparation: Urine samples were thawed and vortexed homogeneously. Each 950-µL urine sample was transferred into a glass tube and mixed with 50 µL of internal standard mixture solution (1 µg/mL 13C4-MEP, 13C4-MBP, 13C4- MBzP, and 13C4-MEHP). Then, 5 µL of β-glucuronidase (200 U/mL) and 245 µL ammonium acetate buffer (1 M, pH 6.5) were added to the tube and vortexed in turn. Next, the samples were incubated at 37 °C for 90 min in a shaking water bath and treated with an SPE column. The SPE cartridges were conditioned with 1 mL of methanol, 1 mL of acetonitrile, and 1 mL of phosphate buffer solution (pH 2.0) added successively. Then, 1 mL of urine sample was diluted with 1 mL of phosphate buffer solution (pH 2.0) and added to the SPE column. The cartridges were then washed with 2 mL of formic acid solution (0.1 M) and 1 mL of water. The cartridges were dried under negative pressure. The target analytes were eluted sequentially with 1 mL of acetonitrile and 1 mL of ethyl acetate. The eluent was collected together and concentrated to dry under nitrogen at 55 °C. The dry residue was reconstituted with 200 µL of 1:9 (v/v) acetonitrile–water for the next step of the analysis.
Method Validation: The analytical method was validated to demonstrate the linear range, recovery, limit of detection (LOD), accuracy, and precision. All samples were prepared as described above using blank (or spiked) urine samples. Urine samples with a series of concentrations (2.5, 5, 10, 50, 100, 200, 500, 1000, and 2000 ng/mL standard mixture solutions, including PA, MEP, MBP, MBzP, and MEHP) were prepared to investigate the linear range, recovery, and LOD. The LOD and limit of quantification (LOQ) were calculated using signal-to-noise of 3 and 10 times based on the blank samples, respectively. Samples spiked at three concentration levels (3.91, 7.81, and 15.63 ng/mL) were used to determine the accuracy of the method. Three repetitions at concentrations of 5, 10, and 50 ng/mL were used to determine the intraday and interday precision in one day and the successive three days, respectively.
Application of the Proposed Method: In this study, 84 individual human urine samples were collected in the same area and stored at -80 °C until analysis. The pretreatment and HPLC–MS–MS methods used to analyze these human urine samples were described above.


Figure 2: MS and MSâMS optimization of the representative target analyte: MBzP.
Results and Discussion
Optimization of HPLC–MS–MS Conditions: To obtain specific, accurate, and sensitive results for quantification during method development, the MS conditions were first optimized by direct infusion. Collision energies were optimized to give the greatest fragmentation with the maximum response for each target analyte. The range of the collision energy was fine tuned from 13–16 eV to 16–19 eV because too much collision energy can cause a loss of valuable stable fragment peaks. The best response of each phthalate ester was about 106 for MS spectra and 105 for MS–MS spectra (see Figure 2). The parent ion and daughter ion for each phthalate in the MRM detection mode was chosen and is shown in Table 2.


Figure 3: Optimization of chromatographic conditions: (a) 30-min and (b) 12-min elution times.
The chromatographic conditions were optimized using the instrumentation and column described in the Experimental section of this article. The ratio between the two phases and gradients was investigated using a 30-min separation time and a solvent gradient ranging from 94% mobile-phase A to the final concentration shown in Table 1. The target analytes were significantly separated. However, detection development time should be as short as possible while still meeting the resolution required. Because these phthalate esters (including PA) have a polar group such as -COOH, the proportion of organic phase (mobile-phase B) was increased to elute the chromatographic peaks faster from the C18 column. The whole elution time was shortened to 12 min, the initial ratio of the water phase was dropped to 70% with the same gradient procedure, and the five target analytes were separated significantly (Figure 3[a] and 3[b]). The chromatographic peaks of MBP and MBzP were overlapped when the water phase was below 70%.

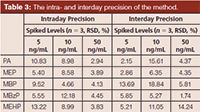
Table 3: The intra- and interday precision of the method.
Method Validation: The calibration curves were obtained using standard solutions of five phthalates with concentrations ranging from 5 ng/mL to 2000 ng/mL. The correlation coefficients are shown in Table 2. The results show good correlation (the correlation coefficient values were greater than 0.99) between the peak areas and the concentrations of the phthalates. The LODs of the five phthalates were 0.85–5.33 ng/mL and the LOQs were 2.82–17.76 ng/mL. The intra- and interday precision of the method are summarized in Table 3. The RSDs of those five phthalate esters ranged from 1.74% to 14.24%. Samples spiked at levels 3.91, 7.81, and 15.63 ng/mL were analyzed three times each. The method validation studies for spiked samples indicated that the present method provides good recoveries and reasonable precision for phthalates at these three levels. As can be seen from Table 4, the recoveries were 81.84% and 125.32% with RSDs between 0.07% and 10.20%. These results demonstrate that the method developed has acceptable precision.
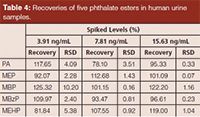
Table 4: Recoveries of five phthalate esters in human urine samples.
Application of the Method: The LC–MS–MS method described above was applied to the analysis of 84 human urine samples collected in one area (Figure 4). Table 5 presents the summary of four phthalate monoester metabolites and PA in these human urine samples. Overall, the detection rates of PA, MEP, MBP, MBzP, and MEHP were 100%, 32%, 99%, 1%, and 21%, respectively. The detection mean concentrations of PA, MEP, MBP, MBzP, and MEHP in all urine samples was 5.31, 16.45, 96.76, 0.07, and 3.83 ng/mL, respectively. The detection concentration ranges of PA, MEP, MBP, MBzP, and MEHP in all urine samples were 1.4–31.3, 0–398.7, 0–663.7, 0–3.1, and 0–184.1 ng/mL, respectively. The results showed that the subjects were exposed to some phthalate esters, especially MEP, MBP, and MEHP, with high concentrations in that region. This study confirmed detection levels of four phthalate metabolites and PA in human urine samples.


Figure 4: Representative LCâMSâMS chromatograms; the blue chromatogram is a nonspiked human urine sample; the red chromatogram is a spiked human urine sample with 200 ng/mL of the five mixture standards.
Conclusions
We developed an LC–MS–MS method for the simultaneous analysis of five phthalate ester metabolites in human urine. The method has proven to be sensitive, accurate, and precise with a relatively short chromatographic run time of 12 min. The results demonstrate that this method can be of value in assessing human exposure to phthalates or other environmental toxicants in environmental toxicology studies.

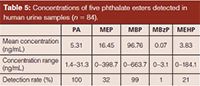
Table 5: Concentrations of five phthalate esters detected in human urine samples (n = 84).
Acknowledgements
This work was supported by funding from the National Natural Science Foundation of China (21107141, 81273478), the Chinese Scientific and Technological Major Special Project (2012ZX09301003-001-010), and the Major Infectious Disease Special Projects (2013ZX10003002-006). Jianhua Cheng and Haijing Li contributed equally to this work.
Jianhua Cheng, Haijing Li, Shengming Wu, Yu Li, Kunpeng Ma, Junjian Fang, Rong Gao, Jiexin Liu, Xianzhong Yan, and Fangting Dong are with the National Center of Biomedical Analysis in Beijing, China.
References
(1) C.R. Blass, Med. Device Technol. 3, 32–40 (1992).
(2) S.Y. Yuan, C. Liu, C.S. Liao, and B.V. Chang, Chemosphere 59, 1295–1299 (2002).
(3) H. Faraji, A. Mirzaie, and S. Waqif-Husain, J. Sep. Sci. 36, 1486–1492 (2013).
(4) R. Rudel and L. Perovich, Atmospheric Environment 43(1), 170–181 (2008).
(5) A. Scott, Chemical. Week 106(28), 13 (2004).
(6) T. Schettler, Int. J. Androl. 29, 134–139 (2006).
(7) G. Latini, Clin. Chim. Acta. 361, 20–29 (2005).
(8) R. Habert, V. Muczynski, A. Lehraiki, R. Lambrot, C. Lecureuil, C. Levacher, H. Coffigny, C. Pairault, D. Moison, R. Frydman, and V. Rouiller-Fabre, Folia. Histochem. Cytobiol. 47, S67–S74 (2009).
(9) H.M. Koch and A.M. Calafat, Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2063–2078 (2009).
(10) E. Samandar, M.J. Silva, J.A. Reidy, L.L. Needham, and A.M. Calafat, Environ. Res. 109, 641–646 (2009).
(11) R. Hauser, J.D. Meeker, S. Duty, M.J. Silva, and A.M. Calafat, Epidemiology 17, 682–691 (2006).
(12) R. Benson, Regul. Toxicol. Pharmacol. 53(2), 90–101 (2009).
(13) L.H. Kembra, V.R. Cynthia, S.W. Vickie, and G.L. Earl, Environ. Res. 108(2), 168–176 (2008).
(14) L.X. Wang and X. Yang, J. Environ. Health 27(3), 276–281 (2010).
(15) P.J. Patyna, R.A. Brown, R.A. Davi, D.J. Letinski, P.E. Thomas, K.R. Cooper, and T.F. Parkerton, Ecotoxicol. Environ. Saf. 65(1), 36–47 (2005).
(16) J.D. Meeker, A.M. Calafat, and R. Hauser, J. Androl. 30, 287–297 (2009).
(17) H. Itoh, M. Iwasaki, T. Hanaoka, H. Sasaki, T. Tanaka, and S. Tsugane, Sci. Total Environ. 408, 37–42 (2009).
(18) K.E. Rakkestad, J.A. Holme, R.E. Paulsen, P.E. Schwarze, and R. Becher, Inhal. Toxicol. 22, 140–150 (2010).
(19) I. Tomita, Y. Nakamura, Y. Yagi, and K. Tutikawa, Environ. Health Perspect. 65, 249–54 (1986).
(20) M. Ema and E. Miyawaki, Reprod. Toxicol. 15(3), 261–267 (2001).
(21) J.J. Heindel and C.J. Powell, Toxicol. Appl. Pharmacol. 115(1), 116–23 (1992).
(22) N. Tatsuhiro, I. Tohru, I. Takeshi, S. Senzo, I. Hajimu, and Y. Takashi, J. Chromatogr. B 780(1), 35–44 (2002).
(23) R. Alzaga, A. Peña, and J. M. Bayona, J. Sep. Sci. 26, 87–96 (2003).
(24) A.M. Calafat, R. Slakman, M. Silva, A.R. Herbert, and L.L. Needham, J. Chromatogr. B 805, 49–56 (2004).

















New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

