Quantitative Metrics to Properly Describe Solute Elution in Size-Exclusion Chromatography


Many different terms and definitions are used to explain the elution and the rate of retention/exclusion/retardation of an analyte in a chromatographic phase system. Size-exclusion chromatography is probably the most challenging chromatographic mode in terms of nomenclature, terms and metrics, with different terms sometimes being used incorrectly. The purpose of this short tutorial article is to review the terms and official nomenclatures for size-exclusion separations and to provide some guidance and recommendations for practicing chromatographers. The interconversion between the different metrics is explained and some examples are presented.
Size-exclusion chromatography (SEC) is unique among the separation techniques, being driven by non-retentive, differential particle pore penetration (solute exclusion). As no interactions with the packing material are required, this chromatographic mode is extremely mild and well-suited for analysis of biological molecules in their native state; as such, it is widely used for antibody-based therapeutic products (1). Size distribution and aggregation are key quality attributes for emerging new modality drugs that are based on increasingly complex polymeric biomolecules and their assemblies (lipid nanoparticles, nucleic acids, viral vectors etc.). Consequently, size-exclusion separations are predicted to be of growing importance and constitute the backbone of quality control testing (2,3). It is thus imperative to use proper quantitative metrics to describe and understand solute elution.
Unlike most liquid chromatography methods, SEC is an entropy-controlled process rather than enthalpy-controlled (4), which necessitates a different approach for an accurate description. In this spirit, one may wonder whether a commonly used term for peak characterization, “retention time”, or its proxy, “retention volume”, (5) are appropriate in a situation where there is not physicochemical retention (ideal SEC separation), and presumably because of this, sometimes, “elution time” can be found instead (6). Neither allows for straightforward communication between system/column comparative studies, which requires further terminology creation. Since efforts to extend the theoretical models in column chromatography to these separations are relatively recent (7), the applied nomenclature is often non-uniform. Hence, the aim for this article is to be a clarifying and unifying guide for SEC practitioners.
Unclear Definitions and Terms: Is There “Retention” in SEC?
It is important to understand the meaning of retention in chromatography in order to accurately describe analyte elution. Historically, volume-based units were used (that is, retention volume, elution volume), but today, time-based units (that is, retention time, elution time, hold-up time) are preferred, simply because time can be directly and intuitively read from a chromatogram. In the case of a non-compressible mobile phase system, time and volume measures are interchangeable and transferable.
Various definitions of retention time can be found in handbooks, guides and articles. Here, we list a few of them: a) “Retention time is the time that a solute spends in a column”; b) it can be defined as the time spent in the stationary and mobile phases”; c) “retention time is a measure of the time taken for a solute to pass through a chromatography column”; d) “retention time is the amount of time a compound spends on the column after it has been injected” (8). Considerations become a bit more complex upon reviewing the definition of retention volume (and/or time) provided by International Union of Pure and Applied Chemistry (IUPAC) (9): “The volume of mobile phase entering the column between sample injection and the emergence of the peak maximum of the sample component of interest, or the corresponding time. It includes the hold-up volume (or time).” Thus, we need to know the meaning of the hold-up volume. The hold-up volume (sometimes referred to as “void-volume” or “dead-volume” and denoted as VM) is defined as: “The volume of the mobile phase required to elute the unretained compound from the chromatographic column” (10,11). Please note that this volume is the sum of the interstitial (external, or interparticle) and pore (internal, or intraparticle) volumes of a column. In SEC, however, the compounds elute before the hold-up volume. Therefore, some chromatographers have delineated “exclusion chromatography” from any form of “adsorption chromatography”. “Exclusion chromatography is based mainly on exclusion effects, such as differences in molecular size and/or shape or in charge. The term “size-exclusion chromatography” may be used when a separation is based only on molecular size” (9,10).
Nevertheless, many chromatographers associate “retention” with physicochemical interactions occurring between a solute and a stationary phase. However, in an ideal SEC separation, molecules do not bind to any component within the column. Therefore, we can quickly run into another problem related to the definition of the “stationary phase”. One of the most common definitions states that “the stationary phase is the part of a column that interacts with the target compound”. This definition suggests that the stationary phase is the “active” part of a column, which is responsible for the adsorptive interactions, and thus, the component that contributes to solute retention. In this manner, it is sometimes referred to as a “bonded phase” (9,10), while the whole solid matter inside a chromatography column is often termed as the “chromatographic bed” or “packing material”.
Due to the ambiguous definitions of “retention” and “stationary phase”, a fundamental question quickly arises: is it correct to say “retention” and “stationary phase” when describing an SEC separation? Many chromatographers agree that neither “retention” nor “stationary phase” are correct terms for SEC. Instead, it is better to simply refer to “elution” and to “column” or “packing material” or “chromatographic bed”. While others (often found in fundamental SEC studies) accept the concepts of retention and stationary phase in SEC, they argue that there is a certain distribution of solutes that are partitioning between the “moving” and “stagnant” phases of the SEC phase system, regardless of the separation mechanism, which is technically accurate. (Please note that here the “moving” refers to the liquid phase–eluent–located in the interstitial column volume, while “stagnant” refers to liquid phase located in the internal pores.) To conclude, both concepts can be applied. The important thing is the selection of a reference point, like “total exclusion”, “total penetration”, or “hold-up” volume/time, so that there is relevance to defining solute elution and/or the migration rate of an analyte.
Measures to Describe Solute Elution in SEC
At any rate, we do not contradict any definition if we simply use “elution time” (te) as a directly readable measure. It refers to the time between the moment of sample injection and the appearance of the peak apex.
The elution of a compound can be expressed in volume units which has the advantage of being independent of flow rate (F). The elution volume (Ve) is the product of te and F:


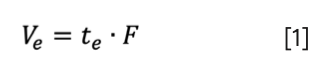
It is even more beneficial to use the ratio of Ve and the empty (superficial) column volume (Vc). Such a “normalized elution volume” (Ve,n) is independent of both flow rate and column dimension. Therefore, this dimensionless number (0 < Ve,n < 1) can be used to compare SEC measurements performed on different column sizes and at different flow rates. The normalized elution volume can be written as:

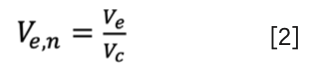
The equilibrium constant related to the partitioning of a solute between the solid and liquid phases is regularly called the “partitioning coefficient”, “equilibrium constant”, “distribution constant”, or “equilibrium partition constant”, and is often denoted as K or KSEC (12,13).

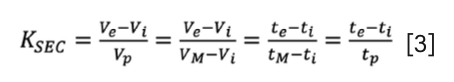
where Vi is the interstitial volume of the column (external porous volume) and ti is the corresponding time, Vp is the internal pore volume of the column (internal porous volume) and tp is the corresponding time, and VM, tM are the column hold-up volume and time, respectively. The value of KSEC ranges between 0 and 1; 0 corresponds to total exclusion, while 1 corresponds to total penetration through the internal pores.
The most used dimensionless unit of solute retention in liquid chromatography is the retention factor (k, k’). The retention factor is a measure of the time the sample component resides in the stationary phase relative to the time it resides in the mobile phase. In other words, it expresses how much longer a sample component is retarded by the stationary phase than it would take to travel through the column with the velocity of the mobile phase (9). Therefore, when analytes are excluded from the pores of a column, they elute with negative retention factors (that is, they only penetrate through a fraction of the column bed). The retention factor is written as:

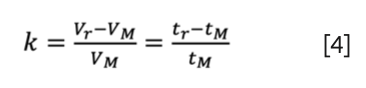
Where Vr and tr are the conventional retention volume and time, respectively. In SEC–formally–Vr and tr can be substituted by Ve and te. The value of the retention factor is -1 < k ≤ 0. Please note that the official symbols of a column’s hold-up time and volume are tM and VM, but in many research papers and handbooks, the hold-up time and volume are denoted as t0 and V0.
According to the USP’s definition, in SEC, “V0 is the retention volume of a component whose molecules are larger than the largest gel pores (11). It may be calculated from the retention time of an unretained compound (t0) and the flow rate:

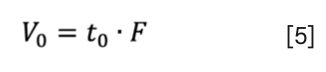
Therefore, V0 is determined by the interstitial column volume (or in other words by the total exclusion volume) and thus V0 = Vi.
Similarly to equation 4, a relative measure of solute elution can be introduced considering the interstitial column volume (or time) as the reference point. This measure is often called the “zone retention factor” (k”), which is an elution factor with respect to the elution time/volume of a non-permeating marker. It expresses the ratio of probabilities of the solute staying in the stagnant mobile phase inside the pores or in the moving mobile phase of the interstitial volume. The zone retention factor is often used when studying bulk, intraparticle, and effective diffusion of a solute. Here, we encourage the readers to formally use the zone retention factor in SEC and call the “elution factor”. The k” is defined as:

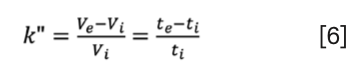
It is worth noting that in SEC, k” is bound by the above, and it reaches its limit (k”max) if a solute penetrates all pores (if Ve = VM then Ve − Vi = Vp):

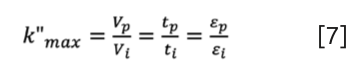
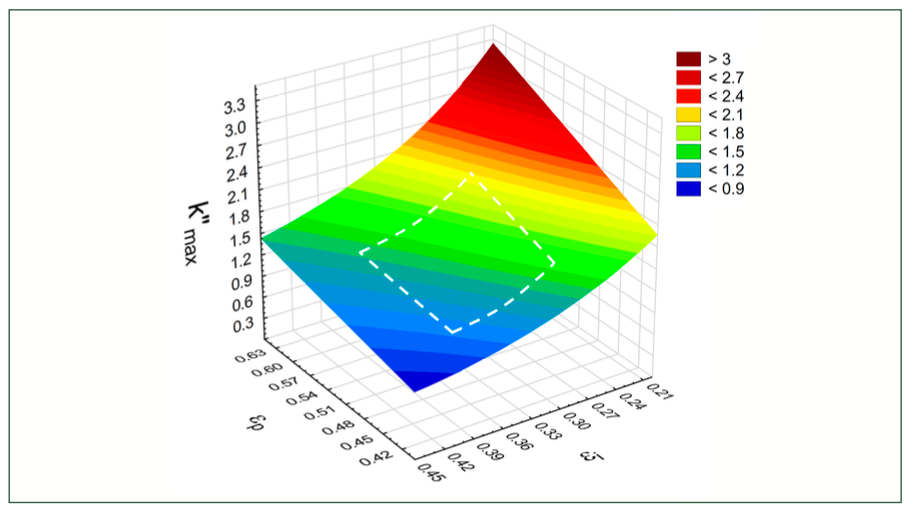
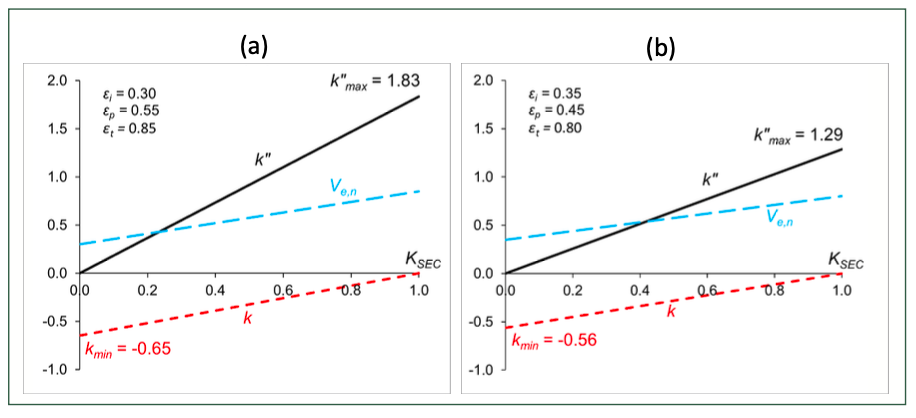
where εp is the pore (internal) porosity and εi is the interstitial (external) porosity of a column. The sum of the two gives the total porosity (εt=εp+εi). It is worth mentioning too, that consequently, analytes elute in a “limited elution window”, as determined by the ratio of pore to interstitial porosity. The k”max is a good measure of the elution window; the higher the k”max, the broader the elution window. A wider elution window provides a higher probability of separating compounds. Figure 1 shows the change of k”max as a function of εp and εi. As expected, the combination of low-interstitial and high-pore porosity results in a wide elution window. The k”max value ranges between 1.1 and 2.1 for most commercial SEC columns.

FIGURE 1: The change of the relative length of elution window (k”max) as function of εp and εi. The area bracketed by the white dashed lines corresponds to the porosity characteristics of most commercial SEC columns.
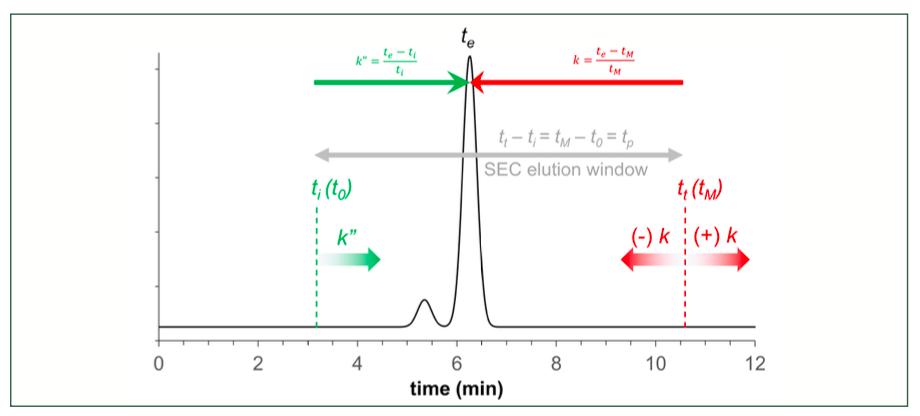
To help practicing chromatographers, Figure 2 shows and explains the directly readable time-based measures on an SEC chromatogram.

FIGURE 2: Illustration of the directly readable time-based units and measures of an SEC separation.
Interconversion of the Different Measures
The various metrics discussed in the above section can be easily interconverted. Some of the important conversions are explained here. When rearranging and combining equation 1 and equation 3, one can express the elution time as the function of porosity, equilibrium constant and flow rate:

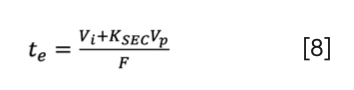
Such a formula is very useful when studying the dependence of te on Vi, Vp or KSEC.
When combining equation 3 and equation 6, a simple conversion between k” and KSEC can be written:

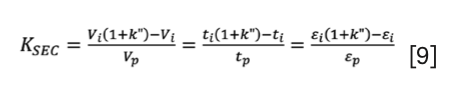
Time-based, volume-based, or porosity-based formulas can equivalently be used.
When combining equation 4 and equation 6, the conventional retention factor (k) can be expressed as a function of k”:

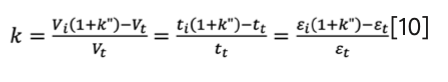
KSEC can be written as a function of k when combining equation 3 and equation 4:

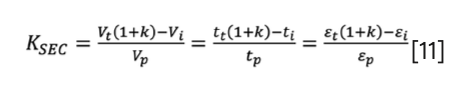
And finally, KSEC can also be expressed from normalized elution volume and column volume by combining equation 2 and equation 3:

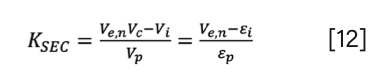
As an illustration, Figure 3 shows the conversion between the dimensionless measures (k, k”, Ve,n and KSEC).

FIGURE 3: k, k” and Ve,n as functions of KSEC for (a) a column with εi = 0.30, εp = 0.55 and εt =0.85, and for (b) a column with εi = 0.35, εp = 0.45 and εt = 0.80.
Column Characteristics Affecting Solute Equilibrium Partition Constant (KSEC)
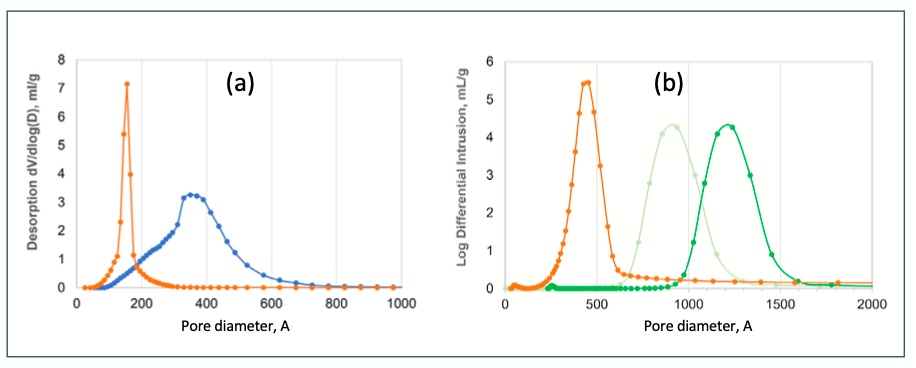
The pore size of the packing material and its distribution play critical roles in SEC separations, since they determine the analyte’s accessibility to the internal pores, and thus the equilibrium constant and the solute’s elution time/volume. Pore volume has a negligible effect on separation of large molecules that are fully excluded from the pores but affect the separation of smaller molecules that can diffuse (penetrate) into the pores.
Pore size, pore size distribution (PSD), and pore volume are often measured by porosimetry that is mostly performed by nitrogen BET and mercury intrusion methods. Based on the physical adsorption of nitrogen on the surface of the packing material at 77 K, nitrogen BET analysis is used to measure mesopores, which covers the pore size range from 2 nm to 50 nm. The Brunauer-Emmett-Teller (BET) equation is used to calculate the pore volume, pore size and its distribution (14). Mercury intrusion porosimetry is used to analyze macropores (>50 nm) and the wider part of mesopores (7.5 nm to 50 nm). The method is based on the penetration of mercury into the pores as a function of the applied pressure. The amount of pressure required for mercury to intrude into the pores is inversely proportional to the size of the pores. The pore size, PSD, and pore volume is calculated from the pressure versus intrusion data by using the Washburn equation, which describes capillary flow in a bundle of parallel cylindrical tubes (15). Please note that the Washburn method gives an estimation, since in reality, pores are not necessarily cylindrical and parallel, but instead possess a complex random and heterogeneous network.
Typical experimental results of pore size and PSD are illustrated in Figure 4.

FIGURE 4: Typical results of pore size, PSD and pore volume measurement of SEC packing materials: (a) Pore volume (V) vs pore diameter (D) obtained from nitrogen BET porosimetry for a narrow (orange) and a wide pore (blue) material, and (b) mercury intrusion porosimetry for a wide (orange) and two ultra-wide pore (grey and green) materials.
Conclusion
Understandably, chromatographers have acquired a range of terms and definitions to explain their separations. A special set of descriptors has arisen for size exclusion chromatography. This form of chromatography solicits the most varied opinions. There are debates on the use of retention, exclusion, or elution, as well as packing material versus stationary phase. That this debate at all exists confirms that SEC is a prevalent approach and that chromatographers are applying their skills and intellectual power. SEC has been an impactful technique for many different fields, ranging from polymer chemistry to biological pharmaceuticals. The value of SEC is evident. Everyone need not agree to using the exact same vernacular; however, if we had our druthers, we might prefer to use the terms “elution time” and “packing material” when discussing SEC. Our preference for these terms over the use of “retention time” and “stationary phase”can be attributed to the years we have spent researching and developing SEC surfaces with weaker and weaker secondary interactions. Accordingly, we are sensitive to connotations of adsorption while discussing idealized SEC behavior. On the other hand, we also recommend using the limit of the zone retention factor (k”max, the ratio of pore and interstitial porosity) as a measure of the width of the SEC elution window, which is an important characteristic of the SEC phase system.
Strong opinions or not, we look forward to more years of advancing the theory and implementation of SEC techniques.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
(1) Goyon, A.; Fekete, S.; Beck, A.; Veuthey, J.-L. and Guillarme, D. Unraveling The Mysteries of Modern Size Exclusion Chromatography - The Way to Achieve Confident Characterization of Therapeutic Proteins. J. Chromatogr. B 2018, 1092, 368–378.
(2) Goyon, A.; Tang, S.; Fekete, S.; et al. Separation of Plasmid DNA Topological Forms, Messenger RNA, and Lipid Nanoparticle Aggregates Using an Ultrawide Pore Size Exclusion Chromatography Column. Anal. Chem. 2023, 95, 15017–15024.
(3) Fekete, S.; Aebischer, M. K.; Imiołek, M.; et al. Chromatographic Strategies for the Analytical Characterization of Adeno-Associated Virus Vector-Based Gene Therapy Products. Trends Analyt. Chem. 2023, 164, 117088.
(4) Striegel, A. M. Size-Exclusion Chromatography: A Twenty-First Century Perspective. Chromatographia 2022, 85, 307–313.
(5) Gritti, F. Resolution Limits of Size Exclusion Chromatography Columns Identified From Flow Reversal and Overcome By Recycling Liquid Chromatography to Improve the Characterization of Manufactured Monoclonal Antibodies. J. Chromatogr. A 2023, 1705, 464219.
(6) Woodall, D. W.; Thomson, C. A.; Dillon, T. M.; et al. Native SEC and Reversed-Phase LC–MS Reveal Impact of Fab Glycosylation of Anti-SARS-COV-2 Antibodies on Binding to the Receptor Binding Domain. Anal. Chem. 2023, 95 (42), 15477–15485.
(7) Chester, T. L. The Combination of Partition, Size Exclusion, and Hydrodynamic Models in Chromatography, and Application to Bonded Phases on Porous Supports. J. Chromatogr. A 2020, 1620, 461011.
(8) Kazakevich, Y. HPLC Theory. In HPLC for Pharmaceutical Scientists. John Wiley & Sons, 2007; pp 25–74.
(9) Maryutina, T. A.; Savonina, E. Y.; Fedotov, P. .S; et al. Terminology of Separation Methods (IUPAC Recommendations 2017). Pure Appl. Chem. 2018, 90 (1), 181–231.
(10) Domínguez, J. A. G.; Díez-Masa, J. C. 2001,73, 969–992.
(11)<621> Chromatography, United States Pharmacopeia, Stage 4 Harmonization (2022).
(12) Giddings, J. C.; Kucera, E.; Russell, C.P.; Myers, M. N. Statistical Theory for the Equilibrium Distribution of Rigid Molecules in Inert Porous Networks. Exclusion Chromatography. J. Phys. Chem. 1968, 72 (13), 4397–4408.
(13) Gritti, F. Theoretical Performance of Multiple Size-Exclusion Chromatography Columns Connected In Series. J. Chromatogr. A 2020, 1634, 461673.
(14) Brunauer, S.; Emmett, P. H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938,60 (2), 309–319.
(15) Washburn, E. W. The Dynamics of Capillary Flow. Physical Rev. 1921, 17, 273.
About the Column Editor

Koen Sandra is the editor of “Biopharmaceutical Perspectives”. He is CEO at RIC group (Kortrijk, Belgium) and Visiting Professor at Ghent University (Ghent, Belgium). He is also a member of LCGC International’s editorial advisory board.

About the Authors

Szabolcs Fekete is a Consulting Scientist in Cell and Gene Therapy, Consumables at Waters Corporation.


Mateusz Imiolek is a Senior Researcher in Cell and Gene Therapy, Consumables at Waters Corporation.


Matthew Lauber is a Senior Director at Waters, where he oversees the development and commercialization of products for the analysis of C> drug products.


Mingcheng Xu is R&D Director of Cell and Gene Therapy, Consumables at Waters Corporation.












How Many Repetitions Do I Need? Caught Between Sound Statistics and Chromatographic Practice
April 7th 2025In chromatographic analysis, the number of repeated measurements is often limited due to time, cost, and sample availability constraints. It is therefore not uncommon for chromatographers to do a single measurement.

Fundamentals of Benchtop GC–MS Data Analysis and Terminology
April 5th 2025In this installment, we will review the fundamental terminology and data analysis principles in benchtop GC–MS. We will compare the three modes of analysis—full scan, extracted ion chromatograms, and selected ion monitoring—and see how each is used for quantitative and quantitative analysis.

Characterizing Plant Polysaccharides Using Size-Exclusion Chromatography
April 4th 2025With green chemistry becoming more standardized, Leena Pitkänen of Aalto University analyzed how useful size-exclusion chromatography (SEC) and asymmetric flow field-flow fractionation (AF4) could be in characterizing plant polysaccharides.

