The Perfect Method, Part II: Where to Start?
Getting started on the right foot is important for efficient method development.
Getting started on the right foot is important for efficient method development.
This is the second instalment in a series on how to develop liquid chromatography (LC) methods in an efficient manner. Last month,1 we considered how to set goals for new methods. This month, we will look at some of the factors involved in selecting a starting point for method development. Although our focus in this series is on method development, in the spirit of LC troubleshooting, we need to remember that many of the choices we make during method development will determine some of the problems that may be encountered or avoided with the final method. So each choice of a specific parameter to optimize should be made with a consideration of what kind of problems might occur during method development and with the completed method.
Playing the Odds
The first choice that we have to make in method development is which chromatographic mode we will use. There are reversed-phase, normal-phase, hydro-philic interaction chromatography (HILIC), ion-exchange, size-exclusion, chiral and other modes from which we can choose. For most of us in the pharmaceutical, environmental and chemical industries, the choice will be reversed-phase LC. I look to Ron Majors' "Column Watch" reviews of the Pittsburgh Conference each spring as a finger in the wind in terms of favoured column technology. Year after year, you'll see that the most common columns, either in terms of overall use or new product introductions, are reversed-phase columns. The reasons are simple — they provide the necessary separation power for a majority of separation problems, are easy to use and are reasonably robust. If I were a gambling man, I'd lay my money down on the reversed-phase bet every time, unless I had a solid reason to choose otherwise.
Some obvious applications require other chromatographic modes. If your sample contains chiral compounds, you need a chiral column, chiral mobile phase, or chiral derivative to enable the separation — reversed-phase LC just won't work. If you need to maintain biological activity of an enzyme or other biomolecule, you will avoid reversed-phase LC because of its strongly denaturing mobile phases. Separation of ionic compounds, particularly inorganic ions, will generally go better with ion-exchange or ion chromatography. The separation of positional isomers is difficult by reversed-phase LC, but generally straightforward by normal phase. So if your samples have special characteristics that preclude use of reversed-phase techniques, use common sense and go with the chromatographic mode that is most likely to lead to success. But for the vast majority of compounds, reversed phase is the best place to start.
Continuous or Discontinuous?
Now that we've decided upon reversed phase as our starting column type, we need to think a bit about the strategy we will use to get a reversed-phase method. There are several variables that we can use during the development process. We need to choose wisely to make the most out of our investment of time and money. One way to classify the parameters is whether they are continuously variable or not, as listed in Table 1. Continuous variables are those that can be changed in infinitely small steps, which gives an advantage in fine-tuning the separation and generally makes them more convenient to use. As the concentration or magnitude of a continuous variable is changed, retention changes in a regular fashion, generally in a linear or logarithmic manner. Discontinuous variables are those that can be changed only in a stepwise fashion, and as a result, retention does not change in a continuous manner. Let's consider the list in Table 1.
Solvent strength: By solvent strength, we mean the amount of the strong solvent in the mobile phase, usually methanol, acetonitrile, or tetrahydrofuran in reversed-phase LC. This is also referred to as percent B-solvent (%B). Of course, we can vary the %B in any increment we want.
Temperature: Temperature can be varied most easily from a few degrees above room temperature to the limit of the column or column oven. This means temperatures in the 30–70 °C range for most systems.
Solvent type: The solvent type can be changed from methanol to acetonitrile to tetrahydrofuran. At first glance, you might think of this as a discontinuous variable, but on closer examination, it is continuous. For example, you can blend methanol and acetonitrile in any combination you desire, making it a continuous variable. In fact, blending solvents can be a very powerful tool so that the characteristics of each solvent can be fine-tuned for maximum separation power.
Additives: The concentration of mobile-phase additives, such as buffers, ion pairing reagents, salts, or amines, can be adjusted in a continuous fashion from not present up to their point of saturation in the mobile phase.
pH: The mobile-phase pH falls in a grey area between continuous and discontinuous variables, so I listed it in parentheses in Table 1. Most reversed-phase columns will operate satisfactorily in the 2 < pH < 8 region, and base-stable columns will operate at higher pHs. The pH can be adjusted in a continuous manner, so in that context, pH is a continuous variable. However, the effect of a change in the pH is not continuous. In the region of ±2 pH units of the pKa of a compound, the pH will modify retention in a predictable and regular manner, but once outside this region, additional changes in pH usually have little effect on retention.



Table 1: Ranking the variables.
Column type: A change in column type, such as C18 to embedded polar phase to cyano to phenyl, comes in discrete steps. For example, you can't move from cyano to phenyl in 1% steps. This discontinuous nature of a change in the column type means that you will not be able to fine-tune this variable. You can have one column or another, but not some fraction of each. There is one company (Bischoff, Leonberg, Germany) that makes a column product that allows the connecting together of discrete column segments containing different stationary phases, but even this is limited to stepwise changes.
Which Variable First?
The ability to fine-tune the effect of a variable and the commercial availability of chromatographic retention modelling software (for example, DryLab, Molnar Institute, Berlin) gives us incentive to focus on the continuous variables of Table 1 before we change column type. Our next decision is which parameter we should focus on first. With many choices, we want to work first with the variable that has a reasonable probability of generating a successful separation. However, at the same time, we want to balance the power of a variable to make a change in the separation with the ease of making adjustments in the variable. That is, we may choose a less powerful variable to pursue first if it is much easier to use than a more powerful one.
I've classified the variables of Table 1 in a slightly different manner in Table 2. I have listed some of the characteristics of the variable that will help us make a decision about which one(s) to choose first. We'll look in more detail at solvent strength, then cover the other variables of Table 2 in less detail, because once the context of Table 2 is understood, most of the information is simple to understand.

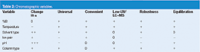
Table 2: Chromatographic variables.
Solvent strength (%B): A change in peak spacing (α) is the desired result of a change in a parameter during method development, so this factor ranks high in selecting our first choice. A change in %B results in a change in α in many instances, but it is not the most powerful variable to elicit a change in peak spacing, so I give it a 0, or neutral rating. A change in the solvent strength works for every compound type and is easy to make — just program a different mobile-phase mixture in the LC system controller — so a + is received for these characteristics. Acetonitrile works well for low-wavelength UV detection (<220 nm); methanol is alright at low wavelengths for isocratic applications, but might not be suitable for gradients; tetrahydrofuran has strong absorbance at <240 nm, but is not widely used. Any of the three solvents will work well for LC–mass spectrometry (MS) applications, although tetrahydrofuran cannot be used when PEEK tubing is present. All in all, detection is not an issue, so another + here. Solvent strength is easy to control and produces robust separations and column equilibration is rapid for both isocratic and gradient experiments, so we get another +. You can see that although solvent strength isn't the most powerful variable to change α, it is positive in all other aspects, so it is usually my first choice in a variable to explore during method development.
Temperature: Temperature is usually considered a weak variable in terms of a change in α, and as a result, many workers ignore it. However, it scores well in all other categories, so it might be worth a more serious consideration in light of other data. For example, as was discussed in the September 2007 "LC Troubleshooting" instalment,1 in some situations, when ionic samples are present, a change in temperature can have the same effect as a change in pH, yet is much easier to control. As we'll see in a later column, temperature and gradient elution is an especially powerful combination of variables for eliciting selectivity changes.
Solvent type: A change in mobile-phase organic solvent from methanol to acetonitrile to tetrahydrofuran can be a powerful way to change selectivity, it works for all types of samples and it is an easy change to make. Tetrahydrofuran has strong UV absorbance below about 240 nm and cannot be used with LC–MS when PEEK tubing is present. Gradients with methanol are difficult below about 220 nm because of baseline drift, but the addition of a UV absorber to the A-solvent can allow use of gradients at lower wavelengths. Column equilibration with acetonitrile and methanol is not a problem, but the use of tetrahydrofuran might take a little more time to equilibrate. Blending different solvents, especially a small amount of tetrahydrofuran with acetonitrile or methanol, will create intermediate solvent properties that can be useful for changing peak spacing. On-line blending of solvents under direction of the system controller can allow exploration of many mixtures in unattended operation.
Ion pair: Ion-pair chromatography is a very useful tool for improving retention, especially for hydrophilic, basic compounds and can be effective to change peak spacing. However, ion pairing does not work for nonionic compounds and has many experimental problems, including very slow column equilibration, so most workers consider other variables before ion pairing is explored.
pH: A change in the mobile-phase pH can be the most powerful variable to change peak spacing, but it only works with ionic compounds. It is not difficult to change the pH, but one must make up a new buffer solution — changing pH by on-line mixing is not reliable. There are buffers that will work well with low UV or LC–MS detection, but many buffers will not work for one or both of these techniques. If the pH is near the pKa of the analyte, the separation can be very susceptible to small changes in pH, such as by use of buffers outside their buffering region, or changes in temperature or organic solvent concentration. However, most separations will be more consistent if the pH of the mobile phase is controlled, because the pH influences the ionization of the column as well as the sample molecules. For this reason, it is best to control the mobile-phase pH, even if the pH is not being explored as a primary variable. In most instances, a 2 < pH < 3 is a good default value for mobile-phase pH unless there is a compelling reason to use another value.
Column type: As mentioned earlier, a change in column type can be a very effective way to change selectivity. This is especially true if one can use specific column selectivity comparison tools (for example, see reference 2) to help choose alternate columns. In the absence of such guidance in selecting a column with "orthogonal" selectivity, the ability to successfully choose a column of different selectivity is limited. For example, there might be more difference between two different C18 columns than between a C18 column and an embedded polar phase column. Because the column is a discontinuous variable, changes are less convenient — the column must be removed and replaced or a column switching valve must be used. And finally, because the typical reversed-phase column costs in the $500 range, the expense of changing a column is much greater than changing any of the other variables. For these reasons, most workers prefer to start with a column that will provide a sufficient number of theoretical plates to separate most sample types, and then change the other variables before changing to a different column type.
On Your Mark, Get Set
If we consider the pros and cons of the various parameters discussed previously, we can choose an intelligent starting point. These conditions can be altered based upon specific sample information, but in the absence of other data, they provide a good place to set your starting blocks in the method development race.
I recommend starting development with a C18 or C8 column that will generate enough theoretical plates for a "typical" sample. This generally means a 150 mm × 4.6 mm column packed with 5 μm particles or a 100 mm × 4.6 mm, 3 μm column operated at 1–2 mL/min. For LC–MS and other applications that don't require quite so much resolving power, a 50 mm × 2.1 mm, 3 μm column operated at 0.2–0.5 mL/min is usually the first choice. A temperature a few degrees above room temperature, such as 30 or 35 μC, is a good starting point. Of course, you should choose one of the newer Type-B or high-purity silica columns and use a new column when starting development of a new method.
Silica-based bonded phase columns are most stable in the 2 < pH < 8 range. Phosphate buffer at pH 2.5–3.0 and 15–25 mM is suitable for UV detection. For LC –MS and other detectors requiring volatile buffers, 0.1% formic acid is a good starting choice. The low pH will suppress ionization of column silanol groups and most acidic sample components. To work above the pKa of most bases will require a speciality column stable to pH > 8, so high-pH operation is usually not the first choice.
Acetonitrile is a good first choice for an organic solvent. It has good UV transparency down to 200 nm and works well with LC–MS. Methanol is a good alternative, but it has stronger UV absorbance at wavelengths below 220 nm. Tetrahydrofuran is less popular because of poor performance at low wavelengths, incompatibility with PEEK and unfavourable handling characteristics.
These column and mobile-phase conditions are a good place to start most separations. Prior knowledge about separations of a particular sample type might suggest other starting conditions. After the starting conditions are identified, the variables of Table 1 or Table 2 can be explored to develop the desired separation.
"LC Troubleshooting" editor John W. Dolan is vice-president of LC Resources, Walnut Creek, California, USA; and a member of the Editorial Advisory Board of LCGC Europe. Direct correspondence about this column to "LC Troubleshooting", LCGC Europe, Advanstar House, Park West, Sealand Road, Chester CH1 4RN, UK.
Readers can also direct questions to the Chromatography Forum at www.chromforum.com
References
1. J.W. Dolan, LCGC Eur., 20(9), 438–443 (2007).
2. L.R. Snyder, J.W. Dolan and P.W. Carr, Anal. Chem., 79, 3254–3262 (2007).



















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


