The Perfect Method, Part 1: What Is Your Goal?
Different methods require different strategies.
Nearly every chromatographer needs to do some kind of method development at one time or another. Whether your job is running a routine liquid chromatography (LC) method that needs an occasional "tweak", you need to develop a one-use method to support chemical synthesis, or you need a robust method to monitor a production process, a good understanding of the principles of LC method development are valuable to know. I have titled this series "The Perfect Method", a little tongue-in-cheek, because, at least in my experience, there is no such thing as a "perfect" method — every method I have seen can always be made better. Herein lies the first principle of method development: "better is the enemy of good enough." You can always make the method just a little better, but it comes at a cost of time that you might not be able to afford. Develop a method that is adequate for the job at hand, then stop.
Over the next several months, we'll look at the subject of LC method development in detail. A few years ago (December 1999–May 2000), I covered this topic with a little different emphasis. In terms of reader feedback, the series was one of the most popular discussions in this column. So we'll look at method development again with a little different twist. Before we start, though, let me caution you that this will not be the final, authoritative treatment on LC method development. If method development is a part of your life in the laboratory, your personal library should include reference 1, which I think is the best book ever written on the subject.
Where Are You Going?
We've all heard Lewis Carroll's quote: "If you don't know where you are going, any road will take you there."
This seems to be the attitude many chromatographers take when they start a method development project. There doesn't seem to be a goal in mind, and even if there is one vaguely formulated, it is felt that a trial-and-error approach will eventually get the job done. Trial-and-error ends up more commonly as error-and-error, which wastes valuable time and money. I think that Laurence J. Peter's take on this subject is much more apropos for method development:
"If you don't know where you are going, you will probably end up somewhere else."
And most of us don't have the luxury of extra time to spend exploring possibilities that lead us away from our goal.
So we need a goal. But that can vary widely. If you desire that method mentioned earlier to use as a quick check of the purity of your synthetic product, a 30 min generic gradient will probably do the job — no need for anything fancy. On the other hand, if your method will need to support a 10000-sample clinical study, the energy spent in reducing the run time from 6 min to 4 min can well be worth the investment. You could think of a number of different criteria that you might use to help define your goals. Here's a list that we use in one of our method development classes at LC Resources:
- Number of samplers
- Run time
- Number of analytes
- Number of matrices
- Sensitivity
- Reproducibility
- Precision and accuracy
- Concentration range
- Qualitative or quantitative
- Equipment or operator limitations
- Sample preparation requirements
- Validation requirements
The list could go on and on. There is no need to discuss each of these criteria in detail, and some will be more important than others for your specific method. Rarely can you answer all the questions, but you can make a good guess in most cases. For example, if you have two active ingredients to quantify in a dissolution experiment at microgram-per-milliliter concentrations, you can be much more specific about your answers than if you are looking at a stability-indicating assay or impurity profile, where force-degraded samples can generate 5–30 peaks, some of which can be in the 0.05–0.1% peak area range relative to the major component. In the latter case, you know that the separation is going to be more challenging than the former, so you can start with an experimental setup that has higher resolving power. A formal document listing the answer to each of the criteria questions might not be required, but it is a good idea to write out a list of as many of the method characteristics as you can think of. If the new method modifies a previous one or is similar to another method, you might be able to use the performance criteria of the existing method as a starting place.
When Are You Done?
You need to have a way to quantify the endpoint of your development efforts so that you don't fall prey to the "just one more experiment" trap that can needlessly extend the method development process. You need some quantitative measurements to go with the qualitative "feel" that your method development is ready to use. One way to do this is to follow the recommendations of regulatory agencies. For example, the US Food and Drug Administration's Center for Drug Evaluation and Research (FDA-CDER) publishes "reviewer guidance" documents designed to help their staff in the review of chromatographic methods for adequate performance relative to the regulations. One of these is a guidance for the validation of chromatographic methods.2 This is not law or policy, but gives us a good idea of what the inspectors will look for in our methods. Four of the quantitative criteria are the retention factor, k, (referred to as capacity factor, k', in the document), tailing factor, Tf, (T in the document), resolution, Rs, and the column plate number, N. These are good measurements to make for the evaluation of any separation, and can form the core of a system suitability test that is run beforerunning each batch of samples with a method.
Retention factor is a measure of the distribution of the sample between the mobile phase and the stationary phase, but from a practical standpoint is another way to measure retention:


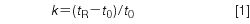
where tR and t0 are the retention time and column dead time, respectively. These are defined as illustrated in Figure 1. The dead time usually is determined either by injecting an unretained substance or identifying the first baseline disturbance in the chromatogram, often referred to as the solvent peak. Retention time is measured from the time of injection to the peak maximum. Ideally, you would like all peaks to be eluted in 2 < k < 10 for the best chromatographic performance, but 1 < k < 20 is acceptable, especially for more complex samples. With k < 2< peaks can be poorly resolved from the unretained material at t0 in most chromatograms, and retention is more sensitive to small changes in the mobile phase composition than when k > 2. The FDA (2) recommends k > 2. For Figure 1, t0 = 1.00 min, tR1 = 3.00 min, and tR2 = 3.40 min, so k1 = (3.00 – 1.00)/1.00 = 2.00 and k2 = 2.4.


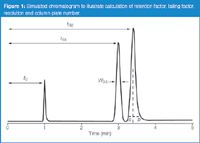
Figure 1
Tailing factor is sometimes referred to as asymmetry factor (with a slightly different method of calculation), and measures the amount that a peak fronts or tails:

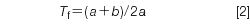
where a and b are defined as shown in Figure 1. A vertical line is dropped from the peak apex and the front and back half-width of a peak at 5% of the peak height are measured. The FDA (2) recommends Tf < 2, but you will have better looking chromatograms, improved quantification, and fewer problems separating minor peaks from major ones if you target Tf ≤ 1.5. For peak 2 of Figure 1, a = 0.10 min and b = 0.16 min, so Tf = (0.10 + 0.16)/(2 × 0.10) = 1.30.
Resolution measures the separation of two peaks in a chromatogram:

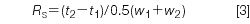
where t1 and t2 are the retention times of peak 1 and peak 2, respectively, and w1 and w2 are the baseline peak widths measured between tangents drawn to the sides of the peak. Determination of the baseline peak width is inconvenient, especially if the baseline is noisy or drifting and if the peaks are not fully separated. Most workers prefer measuring the peak width at half the peak height, w0.5, as illustrated in Figure 1, because it is easier and less error-prone. Now equation 3 becomes

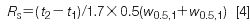
For well-shaped peaks, the valley between the peaks reaches the baseline for Rs = 1.5, but this does not guarantee a complete separation if there is any peak tailing or degradation of the method over time. The FDA (2) recommends Rs > 2. For Figure 1, w0.5,1 = 0.112 min and w0.5,2 = 0.126 min, so Rs = (3.40–3.00)/1.7 × 0.5(0.112 + 0.126) = 1.98.
The column plate number (also called column efficiency) is a measurement of overall column performance. The plate number is influenced most by the packing particle size (smaller particles give larger values of N) and column length (longer columns give larger values of N), as well as many other less important factors, such as flow rate, temperature, mobile phase composition, sample molecular weight, and so forth. The plate number is calculated as follows:

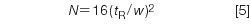
but, as with the measurement of peak width for resolution, it is easier to measure the width at half the peak height, so most workers prefer to use

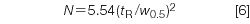
A new 150 mm column packed with 5 mm diameter particles or a 100 mm, 3 mm column will generate N = 12000 or more with an well-behaved test compound, but more in the range of N = 10000 for real samples. The FDA (2) recommends N > 2000. This could be obtained with a poorly performing 50 mm, 5 mm column, so in my opinion, this criteria is not worth much in terms of evaluating the quality of the column. For peak 1 of Figure 1, N = 5.54(3.00/0.112)2 = 3975. One thing to keep in mind is that equations 5 and 6 are for isocratic separations; they will not work for gradient conditions.
Now You Are Ready to Start
You have made a list of the requirements of your method. You have both qualitative (look and feel) and quantitative (Rs, k, run time, and so forth) criteria that you can use to determine if the method is satisfactory. In other words, you know where you are going. In the next instalment of this series, we'll look at how to get to that goal. It really is quite simple, again as stated by Lewis Carroll, "Begin at the beginning and go on until you come to the end: then stop."
"LC Troubleshooting" Editor John W. Dolan is vice-president of LC Resources, Walnut Creek, California and a member of LCGC Europe's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting," LCGC Europe, Advanstar House, Park West, Sealand Road, Chester CH1 4RN, UK.
Readers can also direct questions to the Chromatography Forum at http://www.chromforum.com
References
1. L.R. Snyder, J.J. Kirkland and J.L. Glajch, Practical HPLC Method Development, 2nd ed. (Wiley, New York, 1997).
2. Reviewer Guidance: Validation of Chromatographic Methods, FDA-CDER, November 1994. (http://www.fda.gov/cder/guidance/index.htm).














SPE-Based Method for Detecting Harmful Textile Residues
January 14th 2025University of Valencia scientists recently developed a method using solid-phase extraction (SPE) followed by high-performance liquid chromatography coupled to high-resolution mass spectrometry (HPLC–HRMS/MS) for detecting microplastics and other harmful substances in textiles.





