Paper, Paper Everywhere but None of it Controlled?
LCGC Europe


One of the common threads in the six data integrity guidance documents published to date is the need to control any blank forms used in regulated GXP laboratories. This month’s “Questions of Quality” is focused on how to interpret the regulator’s requirements for this topic. We also pose the question: Is paper the best way to record regulated data?
Chris Burgess1 and Bob McDowall2, 1Burgess Analytical Consultancy Ltd, Barnard Castle, Co Durham, UK, 2R.D. McDowall Ltd, Bromley, Kent, UK
One of the common threads in the six data integrity guidance documents published to date is the need to control any blank forms used in regulated GXP laboratories. This month’s “Questions of Quality” is focused on how to interpret the regulator’s requirements for this topic. We also pose the question: Is paper the best way to record regulated data?
Data integrity is the hottest topic in regulated GXP laboratories today. We have seen how chromatography data systems have been used in falsification and fraud (1) and how a laboratory information management system (LIMS) can help support data integrity in a laboratory (2). In this column, we want to turn our attention to a medium that is common in the majority of laboratories - paper. In particular, we will focus on the items that exist at the back of many standard operating procedures or work instructions to help a chromatographer to record the work covered by the procedure - (drum roll please) - the blank form.
This unsung hero of non-compliance is the subject to a number of comments and controls in the following data integrity guidance documents:
- FDA guide to Inspection of Pharmaceutical Quality Control Laboratories (1993) (3)
- MHRA GMP Data Integrity Definitions and Guidance for Industry, second version (2015) (4)
- WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (2016) (5)
- MHRA draft GXP data integrity guidance (2016) (6)
- PIC/S (Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-operation Scheme) Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments (7)
- European Medicines Agency (EMA) Data Integrity Questions and Answers (8)
The PIC/S and EMA guidances were both issued in August 2016.
Why Control Blank Forms?
Why all the fuss about uncontrolled blank forms? It is very easy - if you don’t get the answer you want, the original work recorded on the form can go in the round filing cabinet, waste paper bin, or shredder and you can complete a new form. This can go on ad infinitum until the work is “correct”. OK, this is an extreme example but it is presented to make a point. Most laboratories work ethically and if an out of specification result (OOS) is found, it will be investigated and documented properly.
What Do the Regulators Want?
Let us look at what the various regulatory authorities say about the control of blank forms.
FDA - Take 1
We’ll go back to the future to discover what the FDA said about control of blank forms in 1993. This quotation is taken from the Guide to Inspection of Quality Control Laboratories (3), written after the Barr Laboratories court case and written to help FDA inspectors in the aftermath of the case. Why quote this when it is nearly a quarter of a century old? Quite simply, it is still as relevant now as it was then because many regulated laboratories work in the same way now as they did then. Would you like some free consultancy advice? Read this document.
In section 13 on the topic of laboratory records and documentation, it states (3):
We expect raw laboratory data to be maintained in bound, (not loose or scrap sheets of paper), books or on analytical sheets for which there is accountability, such as prenumbered sheets.
For most of those manufacturers which had duplicate sets of records or “raw data”, nonânumbered loose sheets of paper were employed.
The FDA expected that if blank forms are used they needed to be prenumbered “for which there is accountability”. Nothing further was offered and laboratories were left to interpret this for themselves. Most laboratories ignored this and nothing changed.
MHRA - Take 1
Moving forward to this century we come to the first data integrity guidance published by the MHRA in 2015. In the section on Designing Systems to Assure Data Quality and Integrity we come upon this regulatory gem (4):
Systems should be designed in a way that encourages compliance with the principles of data integrity. Examples include:
- Control over blank paper templates for data recording
Well that was informative! Even less information and guidance than the FDA.
World Health Organization
In the recent final guidance from the WHO, there is a brief requirement for blank forms (5):
Good document design, which encourages good practice: documents should be appropriately designed and the availability of blank forms/ documents in which the activities are recorded should be ensured;
Good luck to you if you can make much sense of this. All this appears to say is good design of the blank form and its availability needs to be controlled, but we could be wrong.
MHRA - Take 2
Recently in July 2016, MHRA issued a draft GXP data integrity guidance; for comment, the period for this runs until 31st October (6). This document is an update of the GMP guidance but has been expanded to include GCP and GLP - areas that MHRA also regulate in the UK. The section on blank forms have been expanded to state:
“Free access” to blank paper proformae for raw/source data recording should be controlled where this is appropriate. Reconciliation may be necessary to prevent recreation of a record.
An improvement on the first data integrity guidance but not exactly illuminating. The inclusion of the great get out of jail free clause “where this is appropriate” does not help matters - in our view you are either going to control blank forms or you are not.
FDA - Take 2
A new data integrity guidance appeared in 2016 from the FDA (9). This was unusual in that the format took a form of question and answer approach. Bob’s Focus on Quality column in Spectroscopy this month has reviewed this document in more detail (10), however, one question that is pertinent to our discussion is question 6, which asks, “How should blank forms be controlled?”
The answer is much more specific than any guidance that we have looked at so far:
- There must be document controls in place to assure product quality (see §§ 211.100, 211.160(a)).
- FDA recommends that, if used, blank forms (including, but not limited to, worksheets, laboratory notebooks) be controlled by the quality unit or by another document control method.
- For example, numbered sets of blank forms may be issued as appropriate and should be reconciled upon completion of all issued forms. Incomplete or erroneous forms should be kept as part of the permanent record along with written justification for their replacement (for example, see §§ 211.192, 211.194).
- Similarly, bound paginated notebooks, stamped for official use by a document control group, allow detection of unofficial notebooks as well as any gaps in notebook pages.
Now were are getting somewhere. The FDA has reiterated its 1993 stance that blank forms have to be prenumbered, they should be issued to individuals, and after they have been completed there needs to be a reconciliation process that all forms are returned. You must count them out and count them back. If one is incomplete or there are errors made, there needs to be a written justification for issuance of a new form. The returned form is not placed in the round filing cabinet but retained as a GMP record. In addition, there need to be document controls such as design, approval, and secure storage of the master template. A giant leap for regulated mankind?
PIC/S Guidance
One of the most recent guidance documents issued (7) has section 8 focused on the structure of the quality management system (QMS) and the control of blank forms, templates, and records. We suggest that you read the whole section, especially sections 8.4 and 8.6 on expectations for control of forms and records and how to complete records. Two of the principles in this document are:
- The process for generation of working copies of documents for routine use, with specific emphasis on ensuring copies of documents, for example, standard operating procedures (SOPs) and blank forms are issued and reconciled for use in a controlled and traceable manner.
- Guidance for the completion of paper based documents, specifying how individual operators are identified, data entry formats, and amendments to documents are recorded.
There is a lot more information than we can summarize here in the PIC/S document but the principles are included in our proposal for management of templates and forms in this column.
EMA Data Integrity Q&A
This is a web publication that was issued in August 2016 (8) and question 14 is relevant to our discussion: How should the company design and control their paper documentation system to prevent the unauthorized re-creation of GMP data?
In essence there is the requirement to control:
- The template master - this needs to be authored and approved and contain a master reference.
- For loose leaf template forms there needs to be a distribution date, a sequential issuing number, a number of the copy distributed, the department name where the blank forms are distributed to, etc. Each form needs to be traced and accounted for.
- Distributed copies should be designed to avoid photocoping either by using a secure stamp, or by the use of a paper colour code not available in the working areas. Alternative systems would need to be justified.
Like a guilty person who can feel the noose tightening around their neck, this is regulatory payback for poor practices and falsification. Both the PIC/S and EMA guidance documents provide much more detail for blank form control (7,8).
Quo Vadis Blank Forms?
We have made a start with determining what we need to do to control blank forms but this is not enough. We need to work on this some more to fully understand the ramifications of controls required for blank forms and why it is not a good idea to use them. We will look at this in two phases:
- Control of the master template through the phases of design, review, approval, and secure storage
- Use of the pre-numbered form created from the master template in a laboratory to record regulated work
Control of the Master Template
We will present the controls required for blank templates in two phases, firstly by developing and controlling the master template and secondly how it should be used. The way this will be presented in the two figures that follow is as cross-functional process flow diagram. This has the following functions that need to interact in order to control master templates for each blank form and use them in a compliant manner:
- Data governance
- Quality
- Generation
- Distribution
- Completion
- Review
Not all six functions are needed for either of the two phases of the work but all are required across the two phases when looked at holistically. The overall process of controlling the master template is shown in Figure 1.


Figure 1: A process for controlling a master template for a blank form.
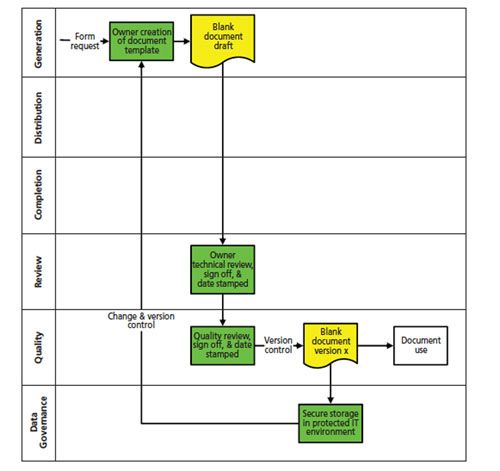
- Each blank form should have an owner allocated to it. This will typically be the subject matter expert (SME) of the overarching standard operating procedure, analytical procedure, or work instruction that controls the use of the form. The form will be designed to accommodate the work and collect records according to the applicable process.
- This form can be designed using a word processor or even a spreadsheet, however, the template must have the name and the version number of the form embedded into it as well as the procedure number to which the form relates. One point that must be ensured is that there is sufficient space for an analytical chemist to enter a value or result.
- When complete the form needs to be reviewed by a different person to ensure that the form is complete, correct and accurate, and matches the requirements of the controlling procedure. If changes are required, the form is returned to the author for update.
- When technical reviews are complete, there needs to be a quality function review and approval and when approved, the date of approval is added to the master form.
- The form should be signed either by hand or by electronic signature.
- Assuming that no changes are required, the master template now needs to be stored securely either in an IT environment on the network or in an electronic document management system with restricted access to it.
At the end of this first process we now have a blank template that is version controlled and under secure storage.
To ensure that only the current version of the template is used there needs to be an effective process for the withdrawal of the old template and replacement with the new version. The quality function needs to maintain an overarching index of all the blank templates, for example, title, reference number including version number, release date, applicable procedure, and storage location.
Use of the Blank Template
Now we come to the issue of how to use a blank template. Instead of printing or photocopying a blank form when you want to do some work, we now need to have a formal process for issuing a controlled and numbered version of the document. The whole process is shown in Figure 2 as a cross-functional process map involving the same functional groups as Figure 1.
- The process starts with a request made to the person or group who manages the issue and reconciliation of blank forms. This function will be outside the laboratory, and will typically be a quality assurance role to ensure independence of operation. An authorized analyst will request a specific form from the document controller who will issue a uniquely numbered version of the blank form created from the current version of the template.
- The unique number is entered into a track and trace system and the name of the requester is entered along with the date and time of issue. This track and trace system in its simplest form is a bound logbook with numbered pages and the entries handwritten by the document controller with information such as date of issue, unique form number, and the person or department to whom the form was issued to.
- The distributed copies of these blank forms should be designed to avoid photocopying either by the use of a secure stamp and ink colour, or by the use of coloured paper not available in the laboratory. An electronic system that can issue forms with a unique number as well as a copy number may be an alternative, but stringent computer validation needs to ensure that this process is secure and only one copy is allowed to be printed. Otherwise a controlled copy will only be issued on paper because an electronic version could be reprinted (the same as blank forms today).
Do you really, really want to do this? You can see the compliance by reading the text or by looking at Figure 2.

Figure 2: Use of a controlled blank template.
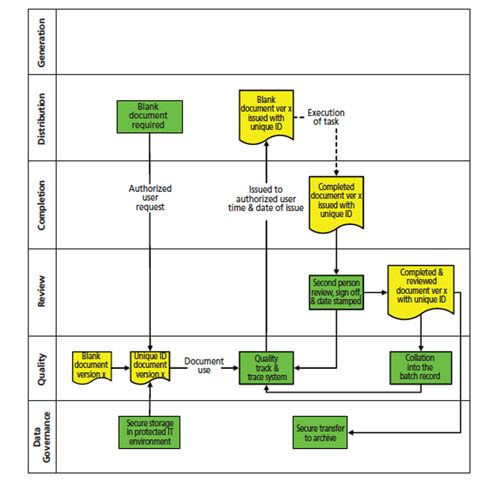
- The forms will be used in a regulated laboratory and completion of them should follow good documentation practices, for example, contemporaneous with the work being executed, completion with an indelible pen, and entries that need to be corrected must be done without obscuring the original entry, and then initialled and dated along with the reason for change. Any blank areas not used need to be struck through, initialled, and dated. Users must not use ditto marks. Do not use a date stamp. Inspectors will now check to see that the person who filled out the form was actually on site when the form was claimed to be completed if falsification is suspected.
- If there is a mistake and a new form is needed, then the form should be returned to the document controller, but before a new form is issued there needs to be a documented rationale for why the form has to be replaced. Get your excuses and grovel pads at the ready for the document controller! The old form must be retained and a new form issued.
- At the completion of the work a reviewer needs to check that the work has been completed correctly and if any calculations are included on the form, they need to be verified as correct - including rounding.
- The form along with other documented evidence is collated into the batch record and the form is reconciled with the track and trace system.
Are Paper Records the Best Way Forward?
As you can see from the processes outlined in Figure 1 and Figure 2, these are far more complex that just using a blank form as we have done previously. However, the whole pharmaceutical industry is now picking up the bill for other people’s laxity, mistakes, and falsification. We now come back to our earlier question - is paper the best way to record GMP data? Looking above the answer is no.
BUT…..
The problem is that many of the software applications that are used in regulated laboratories today are ill-equipped to take over many of the functions currently performed on paper. This is either due to an inability to expand from their core functionality or poor compliance features, such as records stored in operating system files or inadequate audit trail functions including review and electronic signatures not on the records that are signed. Suppliers and users need to work together to ensure adequate functionality and compliance features and this will take time.
BUT…...
Even with the best software some activities may still need to be recorded on paper, such as any dilutions made during sample preparation. This work will need to be recorded on paper and manually entered into an application such as a chromatography data system (CDS).
Summary
We have looked at an area of regulatory concern - control of blank forms. We have outlined one way to achieve this in terms of processes but the mechanism (a word processor file or an electronic document management system) is left to the reader. The process is slow and cumbersome and is really a driver for capturing as much data as possible electronically during an analysis to reduce the compliance overhead.
Please remember, just because you have always worked this way does not mean you can continue to work this way.
Footnote
aWith apologies to Samuel Taylor Coleridge and The Rime of the Ancient Mariner.
References
- R.D.McDowall, LCGC Europe27(9), 486–492 (2014).
- R.D. McDowall, LCGC Europe29(6), 310–316 (2016).
- Inspection of Pharmaceutical Qualiy Control Laboratories. Food and Drug Administration, Rockville, Maryland, USA (1993).
- MHRA GMP Data Integrity Definitions and Guidance for Industry 2nd Edition. Medicines and Healthcare Products Regulatory Agency, London, UK (2015).
- WHO Technical Report Series No. 996 Annex 5 Guidance on Good Data and Records Management Practices. World Health Organization, Geneva, Switzerland (2016).
- MHRA GxP Data Integrity Definitions and Guidance for Industry, Draft version for consultation July 2016. Medicines and Healthcare Products and Regulatory Agency, London, UK (2016).
- PIC/S PI-041 Draft Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments. Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-Operation Scheme, Geneva, Switzerland (2016).
- EMA Questions and Answers: Good Manufacturing Practice: Data Integrity. 2016; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/gmp_q_a.jsp&mid=WC0b01ac058006e06c#section9.
- FDA Draft Guidance for Industry Data Integrity and Compliance with cGMP. Silver Spring, Maryland, USA (2016).
- R.D. McDowall, Spectroscopy31(9), in press (2016).
Chris Burgess is the Managing Director of Burgess Analytical Consultancy Limited, Barnard Castle, County Durham, UK. He has over 40 years experience as an analytical chemist working in and for the pharmaceutical industry. Chris has been appointed to the United States Pharmacopoeia’s Council of Experts 2010 to 2020 and is a member of the USP Expert Panel on Validation and Verification of Analytical Procedures.
“Questions of Quality” editor Bob McDowall is Director at R.D. McDowall Ltd, Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column should be addressed to the editorâin-chief, Alasdair Matheson, at alasdair.matheson@ubm.com







What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






