Overcoming Incomplete Peptide Mapping of Antibody Complementarity-Determining Regions with Alternate Digestion Workflows
Peptide mapping of antibodies is an essential method to monitor peptide modifications in antibody lots that could affect the safety and efficacy of the product. Conventional protocols rely on protein digestion using proteases, such as trypsin, before mapping with mass spectrometry (MS). However, trypsin digestion may cause incomplete mapping of peptides, especially those that include highly hydrophobic peptides. Here, we show how pepsin can be used as an alternative and complementary protease for digestion that allows for improved sequence coverage, especially in proteins with highly hydrophobic regions. We also show that using guanidine hydrochloride (GuHCl) post-digestion improves peptide mapping results. Overall, these two methods—pepsin digestion and GuHCl post-digestion—can be used to provide more comprehensive antibody peptide maps, thereby enabling more thorough quality checking of biopharmaceutical products.
Peptide mapping by liquid chromatography–mass spectrometry (LC–MS) has become the method of choice for in-depth characterization of biotherapeutic proteins, such as monoclonal antibodies (mAbs). The method is now commonly referred to as multi-attribute monitoring (MAM) because it enables simultaneous analysis and monitoring of multiple quality attributes in a single experiment, which traditionally would require multiple independent analytical experiments (1). The MAM approach allows scientists to confirm the primary structure of peptides, identify possible post-translational modifications (PTMs), and provide valuable quantitative information for critical quality attributes (CQAs) at the amino acid level (2,3). Biopharmaceutical companies are now increasingly using MAM for current Good Manufacturing Practice (cGMP) release testing (4), and there is an increasing regulatory awareness and expectation that MAM is employed as an analytical tool in biopharmaceutical development (5–7). Generally, the therapeutic protein is digested by a site-specific protease, and the resulting peptides are separated with LC and detected by LC–MS. The most common protease used for this purpose is trypsin, which cleaves after the basic amino acids, arginine and lysine, and several optimized MAM workflows based on trypsin, have been published (8). Lately, automation of MAM workflows has been gaining traction and recent publications have demonstrated good interlaboratory robustness and consistent results compared to the more conventional and laborious MAM workflows (9,10).
Although trypsin works well for the majority of antibodies and other biopharmaceuticals, trypsin digestion, in some cases, results in small peptides, which are too hydrophilic to be retained on the C18 column. Likewise, in this study, we demonstrated that trypsin digestion may result in large hydrophobic peptides, which absorb to surfaces and return no detectable peptides in LC–MS analysis. In particular, antibody sequences that contain several aromatic amino acids may result in highly hydrophobic peptides after trypsin digestion. These hydrophobic peptides can be difficult to recover, making trypsin-based peptide mapping impractical for those antibodies, which can be problematic if the elusive peptide is part of critical sequences of the antibody, such as the complementarity-determining regions (CDRs) that define antibody epitope specificity. CDRs are important for antibody function because this is where the antibody binds to its specific antigen. Therefore, it is crucial to monitor for peptide modifications in the CDR regions of antibodies.
Here, we demonstrate the use of pepsin as an alternative protease to trypsin and the use of guanidine hydrochloride (GuHCl) post-digestion to access difficult and otherwise elusive hydrophobic sequence regions. Both measures can mitigate against challenges in digestion strategies and provide comprehensive peptide maps, including elusive sequences in the CDR. The MAM workflow presented here builds on the simple and automated digestion workflow published previously (9).
Materials and Methods
We utilized an analytical method that involved automated sample preparation using the Smart Digest Pepsin kit (Thermo Fisher Scientific) combined with LC tandem MS (LC–MS/MS)-based peptide mapping. For all peptide mapping analyses, prepared peptides were analyzed using the Vanquish Horizon ultrahigh-pressure liquid chromatography (UHPLC) system and the Acclaim Vanquish C18 column. Mass spectrometric detection was performed using an Q-Exactive plus or Orbitrap (orbital ion trap) Fusion Tribrid high-resolution accurate mass (HRAM) spectrometer (all instrumentation from Thermo Scientific).
Sample preparation and digestion was automated using the KingFisher Duo Prime purification system with 96 deep-well plates, controlled by Thermo Scientific’s BindIt 4.0 software. Digestions were performed using magnetic Smart digestion kits, which use heat (>70 °C) to denature proteins. Next, 200 μg of the sample was added, and a Smart digest buffer was added to a final volume of 200 μL in each KingFisher 96 deep-well plate, which was digested in the KingFisher Duo for 30 min at 75 °C. After digestion, the samples were actively cooled to 5 °C, and 10 μL of 20% trifluoroacetic acid (TFA) and 70 μL of 8 M guanidine hydrochloride (GuHCl) were added to each sample.
A rapid slit seal cover was placed on the 96 deep-well plates, which were then mixed using a thermomixer and transferred directly to the Vanquish Horizon autosampler for peptide mapping analysis. Peptide mapping by LC–MS/MS was done using data-dependent acquisition (see Figure 1).


FIGURE 1: Detailed description of LC–MS method.

LC–MS/MS data were processed using Protein Metrics BYOS software, using the search parameters shown in Figure 2.

FIGURE 2: Details for software search parameters.

Results
Part One: Trypsin Versus Pepsin Digestion
Complete peptide mapping of antibodies is not always feasible with trypsin when the antibody sequence results in small hydrophilic peptides that are not retained on the C18 column or if the peptides contain several aromatic amino acids, which results in the production of highly hydrophobic peptides. As such, mapping of some antibodies misses coverage of the CDR if hydrophobic regions are present when using trypsin digestion methods. This inability to reliably map CDRs is a major analytical challenge for the characterization and QC of antibodies because this region is critical for antibody function and binding.
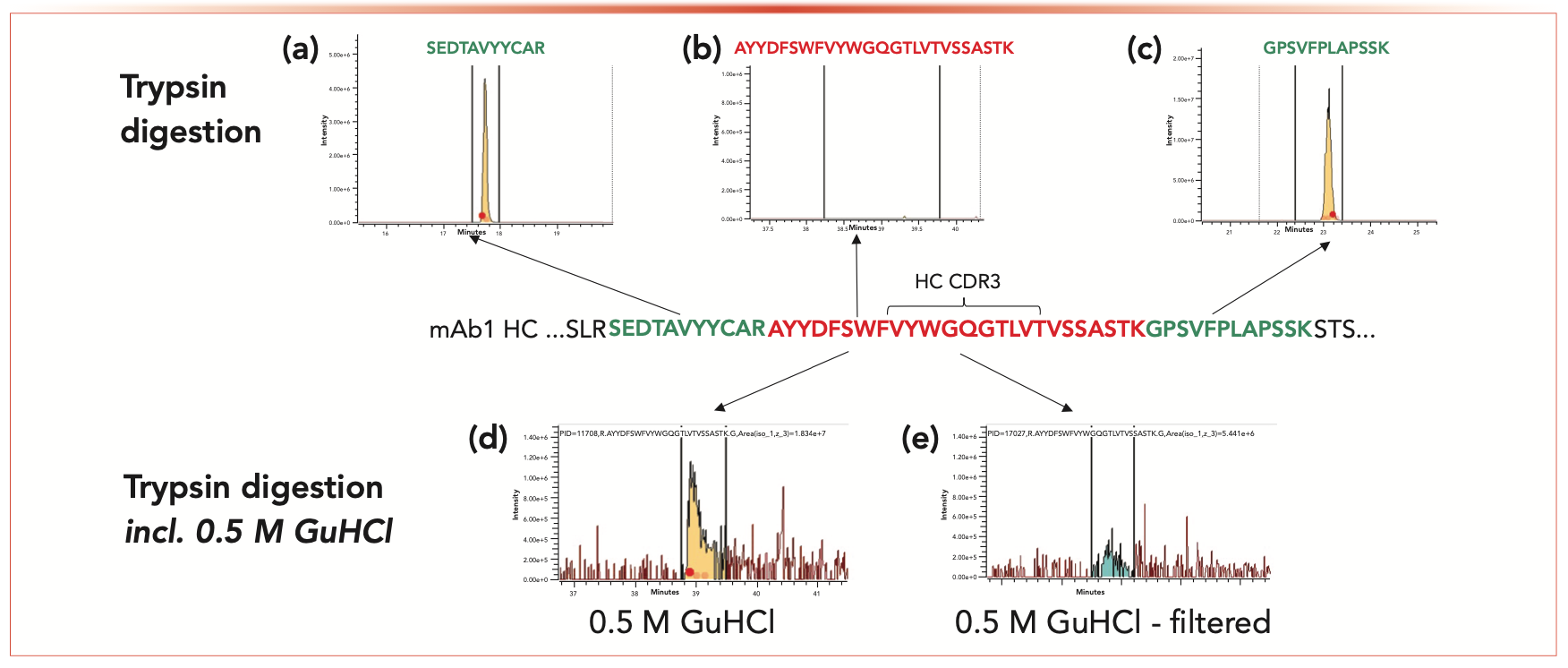
An example is shown in Figure 3, where results of tryptic digestion of mAb-1 showed incomplete mapping of the CDR. It was clear that trypsin was successfully digesting the region (that is, flanking peptides were detected, as seen in Figure 3a), but the peptide spanning the CDR3 region was not detected (Figure 3b). Additives such as urea, GuHCl, dimethyl sulfoxide (DMSO), isopropanol, and acetonitrile that were added during and after digestion did not solve the issue (Figure 3d and 3e).

FIGURE 3: (a,c) Flanking peptides (green amino acids) confirm good trypsin activity; however, the (b) CDR region (red amino acids) was poorly digested. (d and e) Additives like GuHCl (0.5 M) during and after digestion did not solve the issue. For all figures, abscissa label is minutes and the ordinate label is intensity.
Investigating this issue further, we identified other mAbs with the missing sequence coverage in CDRs containing numerous aromatic, highly hydrophobic amino acids. From these findings, we hypothesized that using an alternative protease that digests around the aromatics could create more accessible fragments.
Therefore, we focused on pepsin as an alternative to trypsin. Pepsin digestion resulted in improved sequence coverage in CDRs of all mAbs (see one example in Figure 4b) compared to trypsin digestion (Figure 4a). Figure 4c shows extracted ion currents for peptides from the pepsin digest covering the CDR-3 region, a region which is not covered by the trypsin digestion (Figure 4a).

FIGURE 4: (a) Peptide map showing absence of peptide mapping in the CDR region (marked red) with trypsin digestion, (b) peptide map showing successful detection with pepsin digestion. (c) Example peptide sequences with validated pepsin-based sequences of CDR region. (c) For upper set of three figures, abscissa is labelled minutes, and ordinate is labeled intensity. For lower set of three figures, abscissa is labeled m/z, and ordinate is labeled intensity.
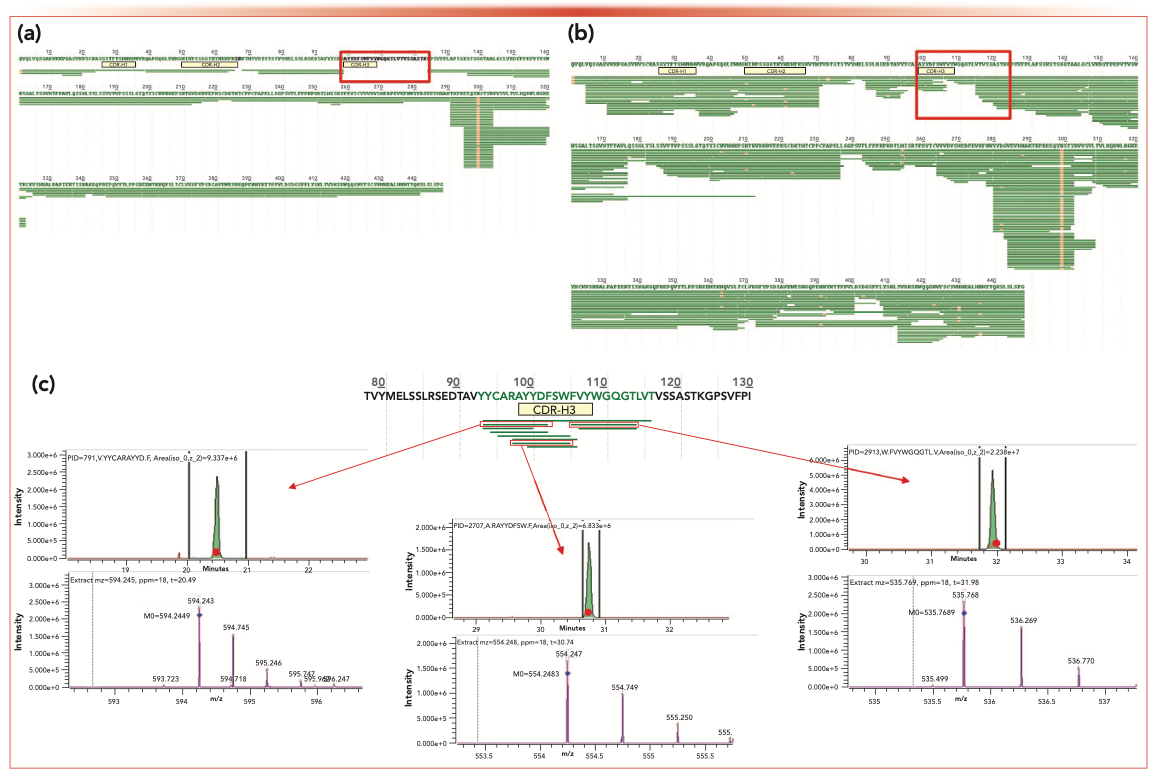
Overall, these results show that pepsin is a viable alternative and complementary digestion method to trypsin for the peptide mapping of antibodies with challenging hydrophobic regions, including those around key regions, like CDRs.
Part Two: Guanidine Hydrochloride Post-Digestion
During our experiments, we noticed a tendency for the hydrophobic peptide sample signal intensity to drop when left for elongated times in the autosampler, which led to the idea that adding GuHCl after digestion could be an effective way of maintaining peptides in a solution and prevent loss of the hydrophobic peptides to the wall of the sample vials. Here, we tested whether post-digestion treatment of samples with GuHCl improved peptide recovery after prolonged storage in the autosampler.
We performed Smart Trypsin digestion of a mAb, subsequently adding 8 M GuHCl to a final concentration of 2 M GuHCl in one of the sample wells directly after digestion. No GuHCl was added to the other mAb digest. The samples were then placed in the autosampler at 5 °C until time of analysis. Samples with 0 M and 2 M GuHCl were analyzed between 1–20 h post-digestion (see Figure 5).

FIGURE 5: Time dependent loss of hydrophobic peptides post-digestion in autosampler; time (minutes) versus total ion current.
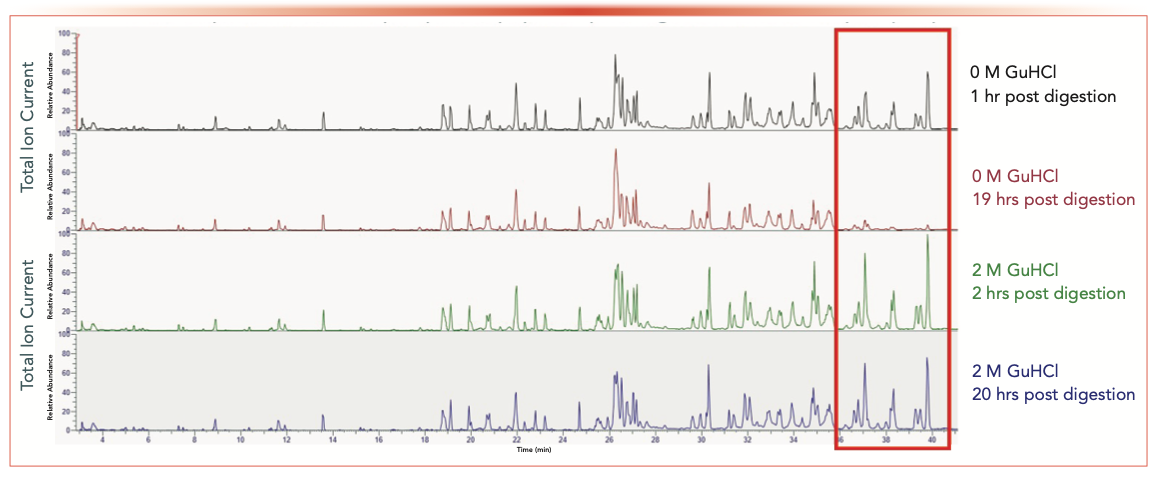
For the sample with 0 M, GuHCl, a distinct loss of hydrophobic peptides is observed for 19 h post-digestion (see red box in Figure 5). In contrast, no loss in signal intensity is observed 20 h post-digestion for the sample with 2 M GuHCl. These results confirm that 2 M GuHCl effectively prevented loss of hydrophobic peptides during storage at 5 °C in the autosampler.
Based on these results, the post-digestion addition of 2 M GuHCl is now a routine part of our peptide mapping workflows for antibodies and other biopharmaceutical products.
Conclusion
Complete sequence coverage by peptide mapping of antibodies is not always possible (for example, when antibodies contain highly hydrophobic regions rich in aromatic residues). There may consequently be poor coverage of CDRs of antibodies when using conventional trypsin digestion methods, which poses an analytical challenge for the characterization and QC of such antibodies.
Here, we show two methodologies that increase the coverage of antibody peptide mapping in chromatography and MS-based workflows: (i) substitution of trypsin for pepsin as a digestion protease increase the coverage of peptide mapping, and (ii) post-digestion addition of GuHCl to prevent time-dependent loss of hydrophobic peptides in the autosampler prior to analysis. These two methodologies improve MS-based workflows, enabling highly accurate and reproducible antibody peptide mapping results. Coupled with the right software tools, high-throughput and ease-of-use capabilities of modern MS systems, the improvements presented here enables laboratories to efficiently screen and characterize challenging mAbs.
References
(1) R.S. Rogers et al., MAbs 7, 881–890 (2015).
(2) K. Diepold et al., PLoS One 7, e30295 (2012).
(3) X.C. Yu, et al., Anal. Chem. 83(15), 5912–5919 (2011).
(4) D. Ren, Trends Biotechnol. 38, 1051–1053 (2020).
(5) S. Rogstad et al., Anal. Chem. 91, 14170–14177 (2019).
(6) I. Apostol et al., Curr. Opin. Biotechnol. 71, 206–215 (2021).
(7) R.S. Rogers et al., AAPS J. 20, 7 (2018).
(8) T. Mouchahoir and J.E. Schiel, Anal. Bioanal. Chem. 410, 2111–2126 (2018).
(9) S. Millán-Martín et al., Anal. Bioanal. Chem. 412, 6833–6848 (2020).
(10) C. Qian, B. Niu, R.B. Jimenez, J. Wang, and M. Albarghouthi, J. Pharm. Biomed. Anal. 198, 113988 (2021).
Ken Cook is the Manager Expert Support Biopharma at Thermo Fisher Scientific, in Hempstead, Hertfordshire, United Kingdom. Dan Bach Kristensen is a Principal Scientist at Symphogen, in Copenhagen, Denmark. Martin Ørgaard and Trine Meiborg Sloth are Senior Technicians at Symphogen. Direct correspondence to: ken.cook@thermofisher.com





 Universe")













New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.
, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

