Optimization of Asymmetrical Flow Field-flow Fractionation (AF4)
LCGC Europe
The technique can be seen as an alternative to size-exclusion chromatography. This article illustrates how to choose parameters such as the dimensions of the separation channel and flow rates.
Asymmetrical flow field-flow fractionation (AF4) can be used on a routine basis to separate high-molecular-weight compounds as an alternative to size-exclusion chromatography (SEC), but the principle of separation is very different and a different approach is required for optimization. This article illustrates how to choose the best experimental parameters. Two optimization parameters are proposed: the time required (TREQ) to separate two compounds that differ by a factor of two in molecular weight and the dilution factor (DILF), which describes the ratio of the actual analyte concentrations during the separation process and after elution from the channel. The TREQ parameter should be minimized to obtain fast separations and the DILF parameter should be minimized when detectability is an issue and the overloading of the separation system is a limiting factor. It is shown that in practice a compromise between these two optimization objectives has to be found.
Asymmetric flow field-flow fractionation (AF4) is a versatile technique for the size-based separation of macromolecules, molecular aggregates, colloids and solid particles. The first experimental study on flow field-flow fractionation by J.C. Giddings et al. was published over 40 years ago,1 but for a long period the development and application of AF4 (or its symmetrical variant) was largely limited to a small number of research laboratories. This was at least partly related to a lack of suitable ready-to-use instruments. In recent years, however, several reliable AF4 instruments have become available on the market and the use of them in routine applications has become more widespread. In the literature several recent reviews can be found on the application of field-flow fractionation in various application fields.2–6



Keys Points
In many laboratories it has been shown that AF4 can be used as an alternative for size-exclusion chromatography (SEC), with a strongly extended molecular-size range. It is typically used for ultra-high molecular weight (UHMW) polymeric materials such as starches and other carbohydrates,7,8 for the study of protein aggregation,9,11 or for the fractionation and characterization of synthetic and natural colloids.12,13 However, the separation principles and characteristics of AF4 and SEC are quite different and optimization strategies and rules-of-thumb for SEC are not always applicable in AF4. In this study, we have tried to derive some rules for the optimization of AF4 separations, starting from the first principles of the technique, and we have validated the outcome with experimental data on the separation of a number of model compounds (proteins).
The Principles of AF4
The principle and experimental set-up for flow field-flow fractionation are shown schematically in Figure 1. The separation in flow field-flow fractionation is performed with a carrier liquid pumped through a flat channel, formed by a spacer between two walls (although a hollow fibre can also be used). In the first (so-called symmetric) systems both walls were porous, and a second pump was used that drove a flow of the same carrier liquid in the perpendicular direction, through both walls of the channel. Macromolecular sample components are retained in the channel by an ultrafiltration (UF) membrane on top of one of the porous walls. Later, Wahlund and Giddings proposed a simplified instrumental system, with only one porous wall, for what was called asymmetrical flow field-flow fractionation (AF4).14 In AF4 the in-going flow (Fin) is split, with the help of flow regulators or an extra pump, in two parts. One part, the channel flow (FOUT), is flushed through the channel in the axial direction towards the detection side outlet. The other part of the in-going flow, the cross flow (FC), passes through the UF membrane and the porous wall. The AF4 set-up requires one pump or flow regulator less than a symmetric system. Commercial instruments use the AF4 principle.

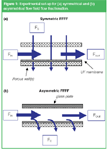
Figure 1
The procedure for a separation by AF4 includes different steps (see Figure 2). First a specific volume of the sample solution is introduced in the channel. This can be done with the inlet flow or through an extra inlet port close to the carrier solution inlet. During and after sample injection a part of the in-going flow enters the channel from the inlet side, but most of the carrier liquid is pumped into the channel from the back end. In this way macromolecules or particles (anything that can not pass the UF membrane) introduced into the channel are concentrated in a thin layer on top of the UF membrane and focused in a thin band close to the inlet port.

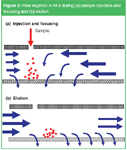
Figure 2
After this focusing step the elution can start. A valve is switched to change the flow regime. The carrier liquid now enters the channel from the inlet side, with a regulated part leaving the channel through the porous wall and the remainder flowing to the detector. The channel flow is laminar (i.e., its velocity is high in the centre of the channel and zero at the walls). Sample components, concentrated by the cross flow in a thin layer on top of the UF membrane, are all flushed with the channel flow towards the detector, but the velocity of a sample band depends on the thickness of the layer into which the particles or macromolecules are concentrated: the smaller the layer thickness of a band, the lower its velocity in the axial direction. The layer thickness for a specific analyte, in the steady state situation after focusing, depends on the cross flow velocity on the one hand and the molecular diffusion of the analyte on the other. Because the cross flow velocity is an instrumental parameter, elution velocity and retention time differences between analytes are purely based on differences in diffusion constant and by that in molecular size.
Experimental
As model compounds β-lactoglobulin (with a MW of 35 kDa), bovine serum albumin (69 kDa), aldolase (160 kDa) and ferritin (440 kDa) were used, all obtained from Sigma Aldrich (Saint Louis, USA). The carrier solution was a 10 mM phosphate buffer at pH 7.4. An Agilent 1100 series degasser and a 1200 HPLC series isocratic pump were coupled with an Eclipse2 AF4 separation system (Wyatt Technology Europe GmbH, Dernbach, Germany) to perform the fractionations. Separation channels with spacers of trapezoidal shape of different dimensions were used and a regenerated cellulose UF membrane with a molar mass cut off of 10 kDa. Samples were injected with a 6-port valve with 10 to 500 μL loops. For detection the UV absorbance was measured at 280 nm with a Spectroflow 757 UV detector (Applied Biosystems, USA).
Sample Injection and Focusing
Analytes to be separated by AF4 have typically low diffusion coefficients and, therefore, enough time should be taken to flush injection loops or autosampler tubing with carrier solution during injection, to transfer the analytes to the separation channel quantitatively. While the sample amount is limited to a certain maximum due to possible overloading effects (see below), the focusing step that is part of the AF4 procedure eliminates any limitation for the sample volume. When samples are strongly diluted a large volume can be injected to improve the detectability. This is shown in Figure 3(a). Here, a certain mass of the model proteins was injected, either as a small volume of a concentrated solution or as a larger volume of a diluted solution. The separation was not significantly affected. In applications with very dilute sample solutions, sample volumes of up to 1 L have been introduced into a normal-size FFF channel.15

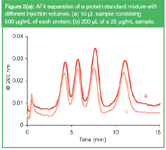
Figure 3a
The focusing time should be optimized for a specific application. When the time allowed for focusing is too short, the sample band in the channel before the elution starts is wider than necessary and the resolution obtained is below optimum. However, when the focusing is long, there is a risk of loss of low-molecular weight material. This is illustrated in Figure 3(b). The recovery of the 35 kDa protein β-lactoglobulin decreases at longer focusing time, even when in this experiment an UF membrane was used with a nominal cut-off value of 10 kDa. The cut-off value of a membrane is an indication for the average pore size of it, but the membrane will contain pores with a wide size distribution. During focusing macromolecules may be lost through the largest pores or through pinholes in the membrane. In practice the focusing is often found to be optimal when the channel volume has been 'swept' 2 to 5 times with the cross flow.

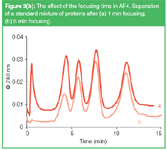
Figure 3b
Retention and Efficiency
The simplicity of the principle of retention in AF4, which relies only on liquid flows and molecular diffusion, makes it easy to predict the relation of the retention of compounds and their molecular size. With some approximations and simplifications of the original treatment,14 the elution time of a compound i can be calculated as:

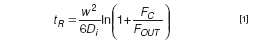
where w is the height of the channel (the thickness of the spacer) and Di the diffusion coefficient of the component. D can be related to size through the well-known Stokes-Einstein formula. Equation 1 shows different crucial aspects of AF4 separations:
- The elution order in AF4 is opposite to that encountered in SEC: small molecules or particles, with a high diffusion coefficient, elute first.
- The retention time of a specific analyte depends on the flow ratio (FC/FOUT). A particular compound can be given any desired retention time by choosing the appropriate flow ratio.
- Retention times do not depend on the length or width of the channel. With a thin spacer elution times will be shorter than with a thicker spacer (at the same flow ratio). Of course, for a fractionation not only the retention times of the analytes are of importance, but also the widths of the peaks obtained. From basic theory (see, for example, reference 16) a simplified formula can be derived for the standard deviation (in time units) σt of a peak of a monodisperse compound with sufficient retention:

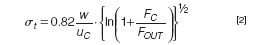
where uc is the cross flow velocity, the cross flow rate divided by the area of the porous channel wall. A remarkable feature of this theoretical formula is that it only contains instrumental parameters. This implies that in a specific fractionation all (monodisperse) compounds elute ideally as peaks with the same width.
While the elution time of a peak depends on the flow ratio (Equation 1), the peak widths depend also on the magnitude of the (cross) flow (Equation 2). For efficient separations the system should be run at flow rates as high as possible. This is illustrated in Figure 4. The model proteins are separated with different flow regimes, using different flow rates but the same flow ratio. The peaks elute at approximately the same time in the different runs, but the resolution between the peaks increases with increasing (absolute) flow rates.

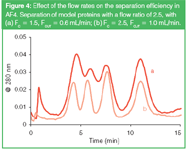
Figure 4
The pressure accompanying a high cross flow velocity will be a limiting factor in practice. As will be shown below, the detectability of the separated compounds is another limiting factor.
Because peak widths do not increase with retention time in a specific fractionation, the efficiency of an AF4 separation can not be quantified with a single plate number. Optimization approaches as used normally in chromatography (e.g., to obtain the highest number of plates per time unit, or to obtain a certain plate number in the shortest time) can not be used in AF4. Therefore, we propose to use another parameter for optimization, TREQ: the minimum time required to get baseline separation between peaks of compounds differing in MW by a factor of 2. A TREQ value then indicates the time required to separate a protein from its dimer, or a 1 MDa narrow polymer standard from a 2 MDa standard. Taking a resolution value of 1.5 as the minimum for baseline separation and assuming that diffusion coefficients are inversely proportional to the square root of the molecular mass of macromolecules (which is a reasonable approximation for, for example, globular proteins or random-coil linear polymers), a surprisingly simple equation can be derived from basic theory that shows the way for the optimization of AF4 separations:

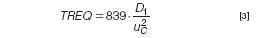
where D1 is the diffusion coefficient of the first eluting compound (the monomer, or the 1 MDa standard in the examples above). The numerical factor in the equation includes the assumed ratio of the diffusion constants and the desired value for the resolution.
Some clear conclusions can be drawn from Equation 3:
- For fast separations the highest possible cross-flow velocity should be applied. The channel flow should then be adapted to obtain the desired resolution (between monodisperse analytes) or selectivity (for samples with a broad distribution).
- Two high-MW compounds, having low diffusion coefficients, can be separated in a shorter time than two low-MW analytes.
- The channel size (length, width, thickness of the spacer) is irrelevant for the separation speed. Of course, with a larger channel a higher cross flow rate is required to obtain the same cross flow velocity.
The cross flow velocity that can be applied will be restricted due to experimental difficulties. At high cross flows a channel may start to leak, low-MW material may be lost through pinholes in the membrane, or sample components may be forced into the pores of the membrane and be irreversibly adsorbed. Such limitations will have to be studied for any specific application.
Overloading and Dilution
When the sample is introduced into the channel, and macromolecular analytes are focused in a thin layer on the accumulation wall in a narrow band close to the inlet, a considerable concentration of the sample components takes place before elution starts. In a typical AF4 experiment an analyte moves through the channel as a band with a volume in the order of 1–2 μL. Therefore, even when the original sample solution can be regarded as 'diluted', during the AF4 process the interaction between analyte molecules may start to play a role. In the highly concentrated bands proteins may spontaneously start to form dimers or aggregates,17 linear polymers can get entangled, and with charged compounds electrostatic interaction becomes important.
When the concentration of an analyte in its band becomes too high, such intermolecular interactions start to affect the elution behaviour, and peaks become wider and distorted. These overloading phenomena are most pronounced with polyelectrolytes (see, for example, reference 18) and ultra-high molecular weight compounds,19 but as Figure 5 shows it can also occur in protein separations. Peak times are shifted and peak widths have increased. The maximum amount of sample (mass, not volume) that can be introduced in the channel before overloading starts to be important depends on the flow regime. With a high cross flow the analytes are more concentrated on top of the UF membrane and overloading is more readily observed.

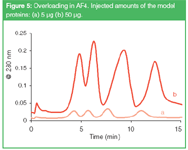
Figure 5
The obvious thing to do when a separation is jeopardized by overloading is to inject less sample. However, the detectability may then become problematic. It should be realized that the concentrated analyte bands that travel through the channel are strongly diluted once they are eluting from the channel towards the detector. The thin layer of solution containing the analytes is merged with the bulk channel flow at the exit port of the channel. We propose to use the dilution factor (DILF) as a parameter to describe these concentrations and dilution processes in the optimization of an AF4 separation. DILF can then be defined as the ratio between the local concentration * of an analyte in the thin band moving in the channel and its concentration in the detector, cdet:

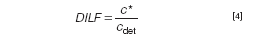
A high DILF value means that one has to choose between two evils. A relatively large amount of sample can be injected, which results in a high true analyte concentration during elution (*), with the accompanying risk of overloading, or a low amount is injected and the detectability (cdet) will be poor. From basic theory (see, for example, reference 18) a simple equation can be derived that describes the influence of instrument and analyte parameters on the dilution factor:

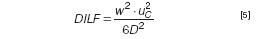
Equation 5 gives some clear indications for the optimization of an AF4 separation:
- High cross flows, as desired for fast separations, will result in a strong dilution and may cause detection problems.
- The use of a thinner channel (spacer) will strongly decrease dilution.
- High-MW (low D) compounds are the most strongly diluted.

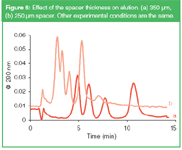
Figure 6
The effect of the spacer thickness on dilution is shown in Figure 6. The model proteins were separated using different spacers in the channel, while the other experimental parameters (flow rates, amount of sample) were kept the same. When the 350 μm spacer is replaced by a 250 μm thick one, elution times are shorter (see Equation 1), and the resolution between the peaks is deteriorated. However, the dilution with the 250 μm spacer is less and sensitivity is improved. [The last peak in the chromatogram of Figure 6(b) can be attributed to the dimer of ferritin; it was not eluted within the data aquisition time window in Figure 6(a).]

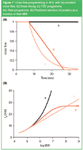
Figure 7
Cross-flow Programming
In many applications of AF4 the components of the sample that have to be separated comprise a wide range of molecular size. For such samples it is difficult to find an optimal cross flow rate. On the one hand a high cross flow is required to obtain enough retention and selectivity for the low-MW sample components. On the other hand, with a high cross flow rate the high-MW sample components will elute very late and very strongly diluted. A valuable tool for the AF4 separation of samples with a high polydispersity is cross flow programming. For this, the cross flow is gradually decreased during elution of the sample components. Different flow programmes can be used in AF4.20 Some programmes are illustrated in Figure 7(a). In the linear decay mode, the cross flow is first kept constant for a certain time and next slowly decreased to zero in a linear way. In the time-delayed exponential (TDE) decay mode, also first the cross flow is kept constant for a certain time, and then decreased exponentially, ideally with a time constant equal to the delay time (the time the cross flow was kept constant). Figure 7(b) shows the effect of flow programming on the expected elution time for globular proteins. It shows that with a constant cross flow the retention time of high-MW compounds becomes impractically long. The size range that can be separated in a reasonable time is clearly wider with the linear decay programming mode. However, at some point (when the cross flow is reduced to zero) all further selectivity is lost. The TDE mode results in a linear relation between the elution time and log MW, with — in theory — no limit on the high side of the size range. An illustration of the power of flow programming was shown in previous work in our laboratory, on the characterization of β-lactoglobulin aggregates.9 The aggregates, that could have MW values well over 10 MDa, had to be analysed together with the remaining monomer (35 kDa). With a constant cross flow the aggregates eluted as a very broad, difficult to quantify peak; with TDE programming the peak was characterized much easier (see Figure 8).

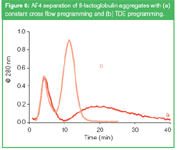
Figure 8
Conclusions
Theoretical considerations and experimental results show that the optimization of an AF4 separation always implies to find the most suitable compromise between separation speed (as quantified with the TREQ value) and detectability (as manifest in the DILF value). The crucial experimental parameter for optimization is the cross flow velocity. The TREQ and DILF parameters are affected in opposite directions when the cross flow velocity is changed.
When speed is the main issue, the cross flow velocity should be chosen as high as allowed by practical limitations (leakage, loss of recovery, adsorption problems). The channel flow can then be chosen to give the optimal compromise between speed and resolution. Of course, with a high cross flow a correspondingly high channel flow will be required for a fast separation. The channel dimensions are not relevant for the separation speed that can be obtained.
When detection sensitivity is an issue and overloading phenomena prohibit to inject simply more sample, it may be necessary to restrict the cross flow velocity to decrease the dilution of the sample components. The dilution can be reduced, without sacrificing speed, by the use of a thinner spacer for the channel. When the available sample amount is not limited, length and width of the channel are also not relevant for the sensitivity that can be obtained. However, in cases when there is only a small amount of sample available (e.g., in biomedical applications), using a smaller channel will improve the detectability. Miniaturization of the AF4 channel will also help to reduce the consumption of carrier liquid.
Furthermore, the power of cross flow programming has been made clear in many practical applications. Flow programming makes it possible to fractionate samples with very wide distributions of molecular mass or particle size.
Rashid N. Qureshi is a PhD student in the Polymer-Analysis Group of the van 't Hoff Institute for Molecular Sciences at the University of Amsterdam. His area of research is in the development and application of flow field-flow fractionation for the characterization and analysis of biomacromolecules.
Wim Th. Kok is a senior lecturer in the same research group, He teaches courses in general analytical chemistry, separation sciences and statistics. His research is focused on the development and application of separation methods (LC, CE and FFF), with an emphasis on miniaturization and on methods for high-MW compounds.
References
1. J.C. Giddings, F.J.F. Yang and M.N. Myers, Science, 193, 1244–1245 (1976).
2. S.K.R. Williams and D. Lee, Journal of Separation Science, 29, 1720–1732 (2006).
3. J. Chmelik, Proteomics, 7, 2719–2728 (2007).
4. B. Roda et al., Analytica Chimica Acta, 635, 132–143 (2009).
5. F. A. Messaud et al., Progress in Polymer Science, 34, 351–368 (2009).
6. S. Cao, J. Pollastrini and Y.J. Jiang, Current Pharmaceutical Biotechnology, 10, 382–390 (2009).
7. G. Modig, P.O. Nilsson and K.G. Wahlund, Starch-Starke, 58, 55–65 (2006).
8. W.J. Kim et al., Bulletin of the Korean Chemical Society, 28, 2489–2492 (2007).
9. R.H. Zhu et al., Analytical Chemistry, 77, 4581–4586 (2005)
10. A. Samontha et al., Journal of Agricultural and Food Chemistry, 56, 8809–8814 (2008)
11. P. Reschiglian and M.H. Moon, Journal of Proteomics, 71, 265–276 (2008)
12. M. Bouby, H. Geckeis and F.W. Geyer, Analytical and Bioanalytical Chemistry, 392, 1447–1457 (2008).
13. S. Dubascoux et al., Journal of Chromatography A, 1206, 160–165 (2008).
14. K.G. Wahlund and J.C. Giddings, Analytical Chemistry, 59, 1332–1339 (1987).
15. H. Lee et al., Analytical Chemistry, 70, 2495–2503 (1998).
16. A. Litzen and K.G. Wahlund, Analytical Chemistry, 63, 1001–1007 (1991).
17. V. Levi and F.L.G. Flecha, Biochimica Et Biophysica Acta-Proteins and Proteomics, 1599, 141–148 (2002).
18. M. Van Bruijnsvoort, R. Tijssen and W. T. Kok, Journal of Polymer Science Part B-Polymer Physics, 39, 1756–1765 (2001).
19. C. Arfvidsson and K.G. Wahlund, Journal of Chromatography A, 1011, 99–109 (2003).
20. K.G. Wahlund et al., Analytical Chemistry, 58, 573–578 (1986).




")





Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.

