Obtaining More Consistent Results
A more practical parameter for system suitability would be to use the peak width for a reference peak...
The topics this month are related to selecting chromatographic conditions that maximize the response for a given compound. This will be of particular interest for workers who run stability-indicating or purity assays for pharmaceuticals, or trace analysis in nearly any application.
Priming Injections
Q: I am running a gradient elution method for the analysis of peptides by LC. I find that it often takes five or more injections of standards before the signal stabilizes sufficiently so that I can run a standard curve. The first injection gives the smallest response, the next a little larger and so on until the response is the same for multiple injections of the same standard. The method comprises a 30 min gradient followed by a 10 min equilibration, so it usually takes several hours before I can run my samples. This obviously wastes a lot of time. Do you have any suggestions?
JWD: The need to make several injections of sample before the signal stabilizes is not uncommon, although some workers can operate for many years without encountering such a method. This phenomenon can be attributed to the presence of (at least) two types of active sites on the column for interaction with the sample molecules. One of these equilibrates much more slowly than the other. These slow-equilibration sites can gradually become saturated with the analyte over several runs, at which time there is no net change of sample on the column with each subsequent injection, so the sample size stabilizes. When the system is shut off, the strongly adsorbed sample might bleed off the column, requiring another round of priming the next time the method is set-up. In other instances, it takes only a single series of priming injections to stabilize the column for all future runs.
As you have noted, priming injections can be time-consuming, especially if the run time is long. In my experience, the important factor is the mass of sample loaded onto the column, not the number of run cycles to equilibrate the column. This implies that there are a couple of techniques that can be used to speed the equilibration process. One would be to make a single high-mass injection to equilibrate the column. An alternative would be to make several smaller injections in a series without running the entire method between each run. In your situation, for example, rather than making five runs to equilibrate, you might make five injections consecutively, then run the gradient just once. Try this — my guess is that you will be able to get the column stabilized much more quickly than your current practice.
Variable Plate Number
Q: One of the system suitability parameters for my method is a minimum plate number for the major peak of interest. This helps ensure that small peaks will be sufficiently narrow that they can be detected properly. However, when I move the method from one LC system to a second system, I can never pass system suitability because the plate number is too low. I have used the same mobile phase, column and sample in an effort to isolate the problem, but still get the same results. The method is a gradient that runs from 10% to 60% acetonitrile with 0.1% trifluoroacetic acid over 15 min on a 150 mm × 4.6 mm, 5 μm particle C18 column at a flow-rate of 1.5 mL/min and a temperature of 35 °C. The sample is prepared in 10% acetonitrile and the injection volume is 10 mL. I also see a shift in retention times of approximately 1 min when I change systems. What can I do to solve this problem?
JWD: I suspect that you are making the common mistake of trying to measure the column plate number N for a gradient method, but using the isocratic calculation. The isocratic formula for N is


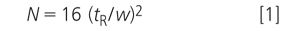
where tR is the retention time and w is the peak width at the baseline. In isocratic separations, the peak width increases as retention time increases and N stays approximately constant throughout the chromatogram. However, this same formula does not work with gradient separations. With gradients, the peak width is more or less constant throughout the run, so you can see that Equation 1 would give larger values of N for larger retention times if the peak width were constant. But we know that N should be relatively constant throughout the run, so the values given for gradients are not correct. Thus, if the peak that you are using for plate number measurements has a different retention time on the two instruments, it will have an apparent difference in plate number, even if the peak width is constant. Although the plate number can be calculated for gradients, the calculation is quite complicated, so it seldom is made. A more practical parameter for system suitability would be to use the peak width for a reference peak — this should stay relatively constant even when the retention time changes, as is the instance in your method.
The shift in retention times that you observe is likely to be caused by a difference in system dwell volume between the two LC systems. The dwell volume is the volume from the point the solvents are mixed until they reach the head of the column. This includes the mixer, transfer tubing, pulse damper (if used) and injector loop for high-pressure-mixing systems, plus the volume of the pump heads for low-pressure-mixing systems. Typical dwell volumes range from 1.5 to 3 mL for high-pressure-mixing systems to 2.5–4 mL for low-pressure mixing systems for general-purpose LC systems currently in production. Systems designed or modified for use with mass spectrometry (MS) detection usually have smaller dwell volumes — in the range of 0.1–0.5 mL, and some older systems can have dwell volumes as large as 5–8 mL. The impact of a change in dwell volume is illustrated in Figure 1. In these simulated runs, everything is the same except the system for Figure 1(a) has a dwell volume of 1.5 mL and that for Figure 1(b) has 3.5 mL of dwell volume. The gradient, as it appears at the head of the column, is overlaid on the chromatogram. The isocratic hold at the beginning of the gradient is a result of the delay caused by the dwell volume. Three general observations can be made when the same method is run on systems with different dwell volumes. First, the time at which the gradient reaches the column differs by the difference in dwell volume divided by the flow-rate (called the dwell time). In the present situation, the difference is 2.0 mL (3.5–1.5 mL) and the flow-rate is 2.0 mL/min, so there is a 1 min difference in the time the gradient reaches the column (arrows in Figure 1). Second, because most gradient conditions are designed so that little or no peak migration takes place under initial gradient conditions, retention times obtained on the two systems will differ by approximately the difference in dwell time. This can be seen by comparing the retention of later peaks in the two runs of Figure 1. For example, the last peak in Figure 1(a) comes off the column at approximately 14 min, whereas the retention time for the same peak in Figure 1(b) is approximately 13 min. Finally, peaks eluted early in the chromatogram often show some degree of migration under the starting gradient conditions, so differences in resolution (or relative retention) might appear when changing systems, especially for the first few peaks in the run. This can be seen with the first two peaks of Figure 1. The resolution, as measured by the valley between the two peaks, can be seen to deteriorate slightly for the system with 3.5 mL dwell volume.

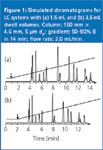
Figure 1: Simulated chromatograms for LC systems with (a) 1.5 mL and (b) 3.5 mL dwell volumes. Column: 150 mm à 4.6 mm, 5 μm dp; gradient: 50â90% B in 14 min; flow rate: 2.0 mL/min.
So how does all this dwell volume business impact practical separations? Because changes in retention, and sometimes resolution, occur when different dwell volume systems are used, the method should be designed with this in mind. For example, system suitability requirements should be sufficiently flexible so that system suitability will pass on different systems that are capable of producing valid results. For Figure 1, one might specify that the first two peaks be eluted between 3 and 5 min and that the resolution must be a specified minimum. This would allow some shift in retention because of dwell volume differences, yet ensure conditions that would produce reliable data. Because dwell volume is such an important parameter in gradient methods, it should be measured for each system and stated in the method. This should allow adjustment of the method from one system to another so that the same results could be obtained. Some of the newer LC systems allow delayed injections, so that the injection can be made after the gradient starts. If this were the situation for the system of Figure 1(b), the method programme could be set such that the sample was injected 1.0 min after the run was started, and the gradient profile and resulting chromatogram would be identical to that of Figure 1(a). Another alternative would be to design the method of Figure 1(a) so that it included 1.0 min of isocratic hold in the program — now the gradient profile and resulting chromatogram would be identical to that of Figure 1(b). The method would be written to adjust the starting conditions so that the dwell volume plus any programmed hold were a constant value for the method.
Peak Height or Peak Area?
Q: Will I get better results from my data if I use peak height or peak area for quantification? It seems like everyone I ask has a different opinion. Can you help me?
JWD: The height versus area discussion has gone on for years, and I doubt that it will go away soon. Unfortunately, the answer to your question is, "it depends." If the peak is large enough that the noise is not significant (e.g., 100:1 signal-to-noise ratio [S/N]), the peak usually will have a smooth transition from one data point to the next. This more often than not is the situation for the output of UV detectors. However, as S/N drops, the importance of noise increases. Also, some detectors give more "ragged" peaks than others. This commonly is the situation for LC–MS–MS peaks, as shown for the peak of Figure 2(a). When I first began to work with LC–MS–MS data, I wondered if there was something wrong when I saw chromatograms like this one, but this is normal. Often, the peak can be smoothed by processing it through an electronic smoothing algorithm or noise filter. The result will be a peak, such as the one shown in Figure 2(b), that is similar to the one that we commonly see with UV detection.

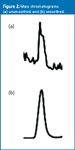
Figure 2: Mass chromatograms: (a) unsmoothed and (b) smoothed.
Now let's consider the precision and accuracy of data for multiple injections of the two samples of Figure 2 when measured by peak height and area. The data system measures the peak height at each point across the peak. When peak height is reported, it is the value of the largest height measurement as a default, but can be the height at a specific retention time. The reported peak area is just the sum of the heights of all the points across the peak, not an area determined from a height and width measurement as one would calculate manually. It should not take much argument to convince you that the area of the peak in Figure 2(a) would give better precision and accuracy than the height. The presence of a single spike in this peak could erroneously report the "true" peak height by 20% or more. The area, or sum of all heights, would average this one spike in with other normal data points, reducing the impact of noise. Alternatively, it is difficult to predict if height or area is better for measurement of the peak in Figure 2(b). Because both height and area are obtained easily from the data system output, I recommend obtaining both height and area values during method development and validation so that the two types of data can be compared. Select the technique that gives the best results and then you will have confidence that you are getting the most out of the data.
Summary
We have examined three problems related to peak size. LC methods might require several priming injections during method set-up so that the peak response stabilizes. Often this is related to the total mass of sample, so you might be able to speed up the process by making one or more large injections rather than making time-consuming runs with a smaller sample load. Be sure to use appropriate system suitability measurements. In the instance of gradient elution, column plate number is difficult to measure, so the use of peak width as a system suitability parameter is both appropriate and convenient. When using an LC method for quantitative analysis, especially at trace levels, you can obtain more consistent results if you use either peak height or peak area. The practical way to make the choice is to make both measurements for a set of data and then select the technique that gives the best results.
"LC Troubleshooting" editor John W. Dolan is vice-president of BASi Northwest Laboratory of McMinnville, Oregon, USA; a training consultant for Rheodyne LLC, the LC Resources Training Group, Of Walnut Creek, California, USA; and a member of the Editorial Advisory Board of LCGC Europe. Direct correspondence about this column to "LC Troubleshooting," LCGC Europe, Advanstar House, Park West, Sealand Road, Chester, CH1 4RN, UK.
Readers can also direct questions to the on-line Chromatography Forum at http://www.chromforum.com
NEXT MONTH in LC Troubleshooting... "Check Valve Problems" — a look at pump design, how check valves work and what steps can be taken to minimize pump problems.

















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

