Nightmare on Lab Street—Are You Haunted by Hybrid Systems?
LCGC North America


Hybrid chromatography data systems are the worst possible solution for any laboratory, whether regulated or unregulated, and yet they are pervasive. Here’s why.
Hybrid CDS applications are pervasive in analytical laboratories. However, they are the worst possible solution because you must synchronize and manage two incompatible data formats: paper printouts and electronic records. We discuss why hybrid systems are the wrong approach and what you can do to remove this specter.
Welcome to a new decade, and a new series of “Data Integrity Focus” articles for your reading pleasure. Let me start by being brutally honest: Hybrid systems are the worst possible solution to have in any regulated or unregulated laboratory. Having made such a bold statement, we will discuss what hybrid systems are and why I have come to that conclusion. I’ll give you some free consulting advice: Don’t use them! As most of you will ignore this advice, I’ll also look at a possible interim solution that could help reduce the volume of paper printed, and make the second person-review of chromatography data simpler and easier.
What is a Hybrid System?
Before we begin to discuss the disadvantages of a hybrid system (and there are no advantages), we had better define what a hybrid system is. The best definition of a hybrid system or approach is found in the WHO guidance on good data management practices:
This refers to the use of a computerized system in which there is a combination of original electronic records and paper records that comprise the total record set that should be reviewed and retained (1).
It then explains in more detail the requirements to link and synchronize the two incompatible media:
An example of a hybrid approach is where laboratory analysts use computerized instrument systems that create original electronic records and then print a summary of the results. The hybrid approach requires a secure link between all record types, including paper and electronic, throughout the records retention period. Where hybrid approaches are used, appropriate controls for electronic documents, such as templates, forms and master documents, that may be printed, should be available (1).
The main points that we can draw out from this definition are:
- A hybrid system has both electronic records and printouts. However, note the phrase “original electronic records.” This is important, because a computerized system, in our case a chromatography data system (CDS), must acquire or create and process electronic records first before they can be printed out; hence they are original e-records that we will discuss later in this article.
- What is missing from the WHO definition is that the analyst and second-person reviewer will sign on the paper printouts as either the performer and reviewer for each analysis, as required by U.S. Food and Drug Administration (FDA) and European Union (EU) regulations (2,3).
- Following on from the last point, there needs to be as secure link between the handwritten signatures on the paper printout with the source electronic records in the CDS.
- There should be, but usually isn’t, controlled copy printing from the system (for example, each printout is labelled copy 1, copy 2, etc.) Otherwise, how sure can we be that the analysis is original, and not a result of testing or integrating into compliance? Alternatively, if there is a second printing, there is a scientifically sound reason for this and copy 1 is retained as part of the complete data for the analysis (2).
Why All the Fuss About Hybrid Systems?
The problem with hybrid systems can be traced back 15 years to the Able Laboratories fraud case. Able was a generic pharmaceutical company based in New Jersey that took a rather unusual approach to analysis, as shown by citation 5 of the Form 483 given at the end of a for-cause inspection:
The substitution of data was performed by cutting and pasting of chromatograms, substituting vials, changing sample weights and changing processing methods…Sample weights were changed by the analyst until a passing result was obtained (4).
The worrying point for the FDA was that they had inspected the company seven times without identifying any falsification of data. The problem was that the inspectors only focused on paper printouts, and never looked at CDS electronic records and audit trail entries-that is, until a whistle blower called the local field office, and the rest is history. This case has resulted in two updates of Compliance Program Guide 7346.832 for Pre Approval Inspections (PAIs), with increased focus on data integrity in both versions (5–7).
Hybrid Records for a CDS
Having established that a hybrid system consists of both electronic records and signed paper printouts, what does this look like for a CDS? Shown in Figure 1 is a hybrid CDS controlling a single instrument. This system can be either a standalone or a networked system, because the principles for a hybrid system are the same regardless of the architecture. The figure is color-coded as follows:
- Yellow indicates that these are paper records that are either generated during the course of analysis or printouts from the CDS application.
- Green is the main flow of data within the CDS from acquiring each data file, processing or integrating peaks, followed by calculating individual aliquot results, and, finally, the reportable value.
- Blue represents the metadata that put the context around the green data files of an analytical run including the audit trail entries.
Seen in Figure 1, the complete record set of a hybrid CDS consists of:


Figure 1: A hybrid chromatography data system.
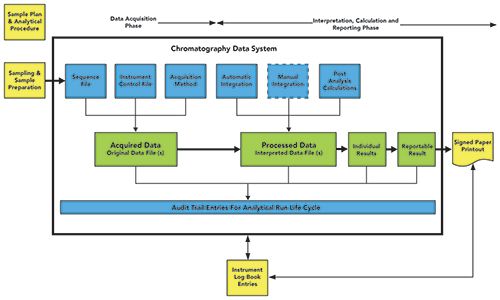
- The electronic records within the data system, including the chromatographic data files and the contextual metadata used to acquire them (sequence file, instrument control file, and acquisition method), then process them to calculate the reportable result (processing method, post run calculations, sequence file containing standard purity and water content, dilutions, and other factors that used to calculate the individual and reportable results.
- Paper records for sampling and sample preparation, as well as the paper printouts from the CDS-this may vary from a summary report to a summary plus all chromatograms. The CDS printout is hand signed by the performer of the test and the reviewer of the whole record set from sampling to final result.
- Entries in the instrument maintenance and use log, that we will discuss later in this article.
You can see that there are two incompatible record formats to manage. It is this combination of electronic records and paper printouts that must be synchronized from creation to destruction throughout the record retention period that makes it the worst situation to be in. Moreover, the WHO guidance goes further and makes the statement that if you look only at paper printouts, poor data management issues and falsification may be missed during the second-person review:
Data integrity risks may occur when people choose to rely solely upon paper printouts or PDF reports from computerized systems without meeting applicable regulatory expectations for original records. Original records should be reviewed-this includes electronic records. If the reviewer only reviews the subset of data provided as a printout or PDF, risks may go undetected and harm may occur (1).
From the perspective of the WHO, it is not looking good for hybrid systems. This statement is also déjà vu of the Able Laboratories fraud case discussed earlier. However, hybrids are very common in many laboratories. But can things get any worse?
In some laboratories, yes, they can, as we will shall see now.
You Cannot Be Serious!
Figure 1 shows the most optimistic version of a hybrid system, where all system suitability test (SST) and sample calculations are performed within the CDS application. Here, all the electronic records for the analysis are contained in a single computerized system, and there is only a single printout from an analytical run.
But...there is always the siren call of the spreadsheet, a ubiquitous application that is easy to use for those lazy sloths that can’t be bothered to read the CDS instruction manual. Furthermore, despite all the suppliers of CDS applications incorporating a wide range of SST parameters and calibration curve models into their software (with the added advantage that you don’t have to print and retype the peak areas into the spreadsheet), the laboratory develops a spreadsheet template to do the same job. I have often wondered whether chromatographers are masochists, and here is the proof.
Now, I suggest you stand back and look at the specter you have created, that which will haunt you unless you change. Instead of the relatively manageable process shown in Figure 1, we now have the stupid (please feel free to insert your own alternative adjective here) situation, where we have made the process more complex, more error prone, with increased record vulnerability, and with not one but two hybrid systems! With transcription error checking between the two systems added for free, besides. I could not write fiction like this.
Chromatographic Data Are Dynamic
Implicit in Figure 1 is the fact that chromatographic data are dynamic and not static records. As you can see, post-acquisition there is automatic and manual integration (if the latter is allowed) to process the data, followed by post integration calculations such as calibration and adjustment by factors such as purity and dilutions. What is the difference between the dynamic and static data? The best description is found in the FDA Guidance on Data Integrity and cGMP Compliance, where an edited version is presented below:
Q1d. How does FDA use the terms “static” and “dynamic” as they relate to record formats?
…static is used to indicate a fixed-data record such as a paper record or an electronic image, and dynamic means that the record format allows interaction between the user and the record content. For example, a dynamic chromatographic record may allow the user to change the baseline and reprocess chromatographic data so that the resulting peaks may appear smaller or larger...(8).
There you have it: Chromatographic data are dynamic records. As such, CDS and the data they contain are firmly in the sights of regulatory authorities worldwide, as slight changes in baseline positioning in a sample chromatogram can turn a failing result into a passing one. These changes can be hidden, if paper is the focus of an audit or inspection.
We will now see what other regulations and guidance documents say about hybrid systems.
EU GMP Chapter 4 Principle
EU GMP Chapter 4 on Documentation includes the following statements concerning hybrid systems:
Many documents (instructions and/or records) may exist in hybrid forms, i.e. some elements as electronic and others as paper based.
Relationships and control measures for…records need to be stated for…hybrid…systems.
Appropriate controls should be in place to ensure the integrity of the record throughout the retention period (9).
Here is the requirement to identify both paper printouts and underlying electronic records for each hybrid system, and to protect both types of records throughout the record retention period. However, the identification of the all records for each computerized system is not always documented in many laboratories, until a data integrity assessment of the system is undertaken.
WHO Good Records Management Guidance
Let me return briefly and brutally to the WHO guidance for the view on hybrid systems. Below are two statements that are about as far as a regulator can go without saying, “Don’t use these systems!”:
The use of hybrid systems is discouraged, but where legacy systems are awaiting replacement, mitigating controls should be in place.
Replacement of hybrid systems should be a priority (1).
How should you interpret “discouraged?” Don’t use hybrid systems would be a good first try.
Record–Signature Linking Requirements
The requirements for linking handwritten signatures on paper to the underlying electronic records in a CDS are found in the 21 CFR 11 regulations for electronic records and electronic signatures, specifically in 21 CFR 11.70:
Electronic signatures and handwritten signatures executed to electronic records shall be linked to their respective electronic records to ensure that the signatures cannot be excised, copied, or otherwise transferred to falsify an electronic record by ordinary means (10).
Now, the problem is that many people think that 21 CFR 11 regulations only apply to electronic signatures as applied to electronic records. However, it explicitly states above that handwritten signatures executed to electronic records shall (please interpret this as must) be linked to their respective electronic records. Therefore, the paper printout must identify not only the key data files that have been used to generate the printout or report, but also the key contextual metadata involved in the generation and processing of the data. Not all contextual metadata needs to be identified on the report (typically, these are the audit trail entries) but if the analytical run is accessed in the CDS, then the remaining metadata need to be easily accessible. This record signature linking is a technical control that is the responsibility of the application supplier. However, the laboratory does not escape responsibility, as there should be a file naming convention for contextual metadata, such as “chromatographic methods” and “processing methods,: and so on.
Why Can’t I Use Paper as My Original Records?
Rather than answer this question, I’ll get my North American advertising agency to answer it on my behalf. Question 10 in the FDA data integrity guidance asks, “Is it acceptable to retain paper printouts or static records instead of original electronic records from stand-alone computerized laboratory instruments, such as an FT-IR instrument? (8)”
The simple answer is no. However, a better discussion on this issue and focused on chromatographic data can be found on the FDA web site under the snappy title of “Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance-Records and Reports (11).” Here, Question 3 asks: “How do the Part 11 regulations and ‘predicate rule requirements’ (in 21 CFR Part 211) apply to the electronic records created by computerized laboratory systems and the associated printed chromatograms that are used in drug manufacturing and testing?”
The FDA starts to answer the question by stating that some people misinterpret the Part 11 Scope and Application guidance (12) (lines 164 to 171) to mean that, in all cases, paper printouts of electronic records satisfy predicate rule requirements in 21 CFR 211. This is not the case, as the guidance also states:
…persons must comply with applicable predicate rules, and records that are required to be maintained or submitted must remain secure and reliable in accordance with the predicate rules (12).
The two applicable GMP predicate rules cited are 21 CFR 211.180(d) and 21 CFR 211.68(b) (2). The former requires records to be retained either as original records or true copies, and the latter states backup data are exact and complete. The FDA view is that:
The printed paper copy of the chromatogram would not be considered a “true copy” of the entire electronic raw data used to create that chromatogram, as required by 21 CFR 211.180(d). The printed chromatogram would also not be considered an “exact and complete” copy of the electronic raw data used to create the chromatogram, as required by 21 CFR 211.68. The chromatogram does not generally include, for example, the injection sequence, instrument method, integration method, or the audit trail, of which all were used to create the chromatogram or are associated with its validity.
Therefore, the printed chromatograms used in drug manufacturing and testing do not satisfy the predicate rule requirements in 21 CFR Part 211. The electronic records created by the computerized laboratory systems must be maintained under these requirements (11).
This principle applies to all GMP records: Do not ignore or delete e-records and only rely on paper printouts. This applies to CDS and spectrometry systems as well as spreadsheets. Therefore, you should now understand the rationale for my comment earlier in this article that a CDS and spreadsheet combination results in two hybrid systems.
Can I Reduce Paper with a Hybrid System?
There is a suggested approach for reduction of paper printouts from hybrid systems that can be found in the WHO data integrity guidance in the Appendix under “Special Risk Considerations for Attributable” (1). A prerequisite is that there needs to be adequate security and backup for the system. As noted earlier, a hybrid approach is likely to be more burdensome than an electronic workflow with electronic signatures. An approach is shown in Figure 2 and described below:

Figure 2: A suggested approach to reduce paper output from hybrid systems (process is from reference (1), figure is from reference (13).
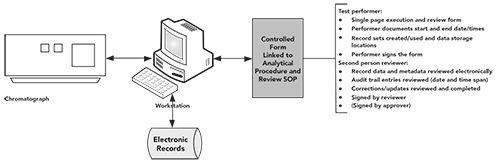
- Create a controlled blank form that is linked to the SOP for the CDS analysis and data review. There needs to be sufficient space for the performer, reviewer, or approver of the analysis to enter the records created or used, as well as hand sign the form
- As the performer executes the analysis, the data files created and the associated contextual metadata used are recorded on the form, along with the location of where the records are stored (for example, in a directory or project). When complete, the performer signs the form to link his or her signature to the CDS electronic records generated and used in the analysis.
- The form passes to the reviewer, who now has a list of electronic records to review, along with applicable audit trail entries. The review is conducted electronically on screen; no chromatograms are printed. The reviewer checks the record set to ensure work has been conducted correctly, and that all calculations are correct. In addition, if technical controls are not in place to control data storage, locations where data could be stored to mask unofficial testing or searches to identify short or aborted sequences should be conducted by the reviewer. When complete, the reviewer will sign for completing the review.
- If required by local procedures, the controlled form is passed to an approver. Note that this is not a regulatory requirement; only two people are required by 21 CFR 211.194(a) (2).
This reduces the volume of paper printed by the system to either the controlled blank form and possibly a simple report of the results. Reiterating the points above, all review is carried out on screen which makes the backup and security of the electronic records in the CDS paramount. The benefit of this approach is that it makes long term retention simpler and easier. However, we have forgotten an essential element in both analysis and review: the instrument log book, as shown in Figure 1.
The Trilogy of Electronic Records, Paper, and Log Book
When using a hybrid system, there are three essential elements to ensure data integrity that are shown in Figure 1.
- The electronic record set for the analysis: the chromatography data files and the associated contextual metadata, such as sequence file, instrument control method, sequence file, processing method and audit trail entries.
- Paper records comprising sample preparation worksheets, including balance records for sample weights and printouts from the CDS after interpretation and calculation of the reportable result(s), or the controlled worksheet described above.
- Instrument log book entries for the analytical run
The instrument log book is an essential component for ensuring data integrity. Log books should record the maintenance, use, calibration, and repair of a chromatograph, as required by 21 CFR 211.182 and EU GMP Chapter 4.31 (2,9). However, where technical controls are either not available or use of them is not feasible (such as where logging off would stop the analysis), the instrument log should record actions of users who interact with the system but are not logged in. This is acknowledged in both the WHO and PIC/S guidance documents (1,14), with the key section of the PIC/S document stating:
some computerized systems support only a single user login ...Where no suitable alternative computerized system is available, equivalent control may be provided by third party software, or a paper-based method of providing traceability (with version control). The suitability of alternative systems should be justified and documented. Increased data review is likely to be required for hybrid systems (14).
The additional effort and time for the review is justified, as it is a procedural control that is error prone rather than a technical control that is validated and enforced by the CDS application software. For those that would like to go into more detail on this topic, I have written more about the role of an instrument log book in a “Focus on Quality” column in Spectroscopy (15). Furthermore, the use of an instrument log book in lieu of an effective audit trail will slow the second-person review process, as noted by Newton & McDowall (16), which is yet another reason for not using hybrid systems.
The Last Word
Ideally, hybrid chromatography data systems should be reconfigured and revalidated to work electronically with electronic signatures. The principles for this design should be:
- Electronic data should be captured at source.
- All work should be done electronically, with all calculations performed within the CDS and report being electronically signed
- Ideally, sample weights and analysis identities should be downloaded from a LIMS or equivalent application. As this process is validated, the only transcription error checks are for manually entered data.
- Transfer of results to a LIMS or similar application should only be performed by a validated and automatic transfer.
- Know where the data are stored, so that electronic records can be retrieved quickly when required (ideally, this should be on a secure and resilient drive on the network that is backed up regularly by the IT function).
The best approach is to have a CDS architecture, even for a single chromatograph, that has the following attributes:
- networked solution for record security and backup
- database to manage records.
For more information on this topic, please read the four-part series on an ideal CDS for regulated laboratories (17–20).
Summary
We have looked at why hybrid systems are the worst possible solution for a regulated laboratory, because there are two incompatible record formats to manage. Focusing on just paper records means that poor data management practices or falsification may be missed, and therefore it takes longer to review hybrid records, because both sets of records plus the instrument log need to be reviewed. If a CDS lacks full audit trail functionality, then a paper record of activities must be maintained in a log book.
In the next article, we will look at audit trail review systems.
References
- WHO Technical Report Series No. 996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016).
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration: Sliver Spring, Maryland, 2008).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control (European Commission, Brussels, Belgium, 2014).
- Able Laboratories Form 483 Observations (2005) (accessed December 23, 2019); Available from: https://www.fda.gov/media/70711/download.
- FDA Compliance Program Guide CPG 7346.832 Pre-Approval Inspections (Food and Drug Adminsitration: Silver Spring, Maryland, 2010).
- FDA Compliance Program Guide CPG 7346.832 Pre-Approval Inspections (Food and Drug Administration: Sliver Spring, Maryland, 2019)..
- R.D. McDowall, Spectroscopy 34(12), 14–19 (2019).
- FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (Food and Drug Administration: Silver Spring, Maryland, 2018)..
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation (European Commission, Brussels, Belgium, 2011).
- 21 CFR 11 Electronic records; electronic signatures, final rule, in Title 21 (Food and Drug Administration: Washington, DC, 1977).
- FDA Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance–Records and Reports (2010) (accessed December 22, 2019); Available from: https://www.fda.gov/drugs/guidances-drugs/questions-and-answers-current-good-manufacturing-practices-records-and-reports.
- FDA Guidance for Industry, Part 11 Scope and Application (Food and Drug Adminstration: Rockville, Maryland, 2003).
- R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
- PIC/S PI-041-3 Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments, Draft (Pharmaceutical Inspection Convention/Pharmaceutical Inspection Cooperation Scheme, Geneva, Switzerland, 2018).
- R.D. McDowall, Spectroscopy 32(12), 8–12 (2017).
- M.E. Newton and R.D. McDowall, LCGC North Am. 36(8), 527–529 (2018).
- R.D. McDowall and C. Burgess, LCGC North Am. 33(8), 554–557 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am. 33(10), 782–785 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am. 33(12), 914–917 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am. 34(2), 144–149 (2016).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com


: Now, Next, and How?")


![[1].jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Ff970d09fd9a78d8d758b609bc5555ac0efcf1f2b-1000x228.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "[1].jpg")

What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






