New Sample Fractionation Strategies for Proteomic Analyses by LC–MS
Special Issues
Mass spectrometry has long been a preferred tool for protein identification and biomarker discovery, but preparation of biological samples remains a challenge. Hindrances include the wide range of protein concentrations, sample complexity, and loss or alteration of important proteins due to sample handling. This article describes recent developments in sample fractionation technologies that are overcoming these challenges in interesting ways and are enabling in-depth proteomic studies that were not possible in the past.
Proteomic analyses based upon liquid chromatography–mass spectrometry (LC–MS) are quite powerful, but when protein digests are too complex, lower-concentration peptides often escape detection. This is where sample preparation plays a key role. For almost any proteomic analysis today, robust and reproducible fractionation techniques are the key to ensuring that MS-MS can identify the maximum number of proteins. At the same time, to avoid inadvertent loss or alteration of critical proteins, it is important that fractionation steps minimize the number of sample manipulations.
Successful fractionation of biological samples is crucial for identification of diagnostic markers and therapeutic targets. Some biological samples are excellent candidates for biomarker study, but researchers have not yet fully explored them because of difficulties with sample preparation. Examples include plasma and serum analyses, in which highly abundant proteins can mask the low-abundance proteins of interest, and membrane-protein analysis, in which hydrophobicity contributes to losses during sample handling. With new developments in sample fractionation techniques, these previously inaccessible samples are becoming accessible.
The Challenge of Plasma
Because plasma comes into contact with almost all of the tissues in the body and is easily obtainable, it is an important source of new protein biomarkers for diagnostics and drug discovery and development. However, plasma is a very complex sample that presents unique challenges, including the fact that proteins are present over a wide concentration range — from micrograms or milligrams per milliliter for the most abundant proteins down to nanograms or picograms per milliliter for the less abundant species. Seven of the most abundant proteins in plasma — albumin, IgG, transferrin, haptoglobin, IgA, anti-trypsin, and fibrinogen — make up about 90% of the total protein mass. These proteins must be removed to enable detection of low-concentration biomarkers by LC–MS.
Another challenge with plasma is the complexity of the sample that remains after removal of the most abundant proteins. Sample complexity is an obstacle that is common to many proteomic samples. The interesting proteins must be fractionated to produce simpler mixtures with fewer coeluted species. Only then is it possible to maximize the number of proteins that can be identified in the search for important protein biomarkers.
Reproducible Removal of High-Abundance Proteins
Multiple affinity removal (immunodepletion) is an excellent method for eliminating highly abundant proteins. With this technique, the sample is passed through a column or other medium, where antibody–antigen interactions remove targeted proteins. Immunodepletion requires optimized buffers that minimize binding of nontargeted proteins, and that ensure reproducibility and long column life. The method requires one buffer to load, wash, and regenerate the column, and a second buffer to elute the bound high-abundance proteins.
Several factors contribute to successful immunodepletion. First and most obvious, the process must be very selective. In other words, it must remove a very high percentage of the undesired proteins while allowing the other proteins to pass through. The removal of abundant proteins can be optimized by using antibodies that are specific to both the protein and the species of interest, and by mixing them in the proper (biological) ratios. Well-designed columns can remove at least 98% of seven targeted high-abundance proteins, as demonstrated by enzyme-linked immunosorbent assays (ELISA).
To maintain high recoveries of biomarkers, it is equally important to minimize the nonspecific binding of interesting low-abundance proteins to the antibodies. The immunodepletion process also must circumvent a tricky problem — the association of many biomarkers with sticky carrier proteins such as albumin. To avoid removing these biomarkers along with the albumin, it is important that buffers minimize protein–protein interactions.
To enable detection of low-abundance proteins by LC–MS, it is very important for the affinity removal column to have sufficient sample capacity. Currently, commercially available 100 mm × 4.6 mm columns for human fluids allow loading of 70 μL of plasma, while 100 mm × 10 mm columns allow loading of 300 μL of plasma. This high loadability permits LC–MS-MS detection of biomarkers in the nanogram-per-milliliter range.
Finally, the multiple affinity removal process should be easily automated, fast, and reproducible. Figure 1 shows an overlay of six chromatograms obtained during the automated removal of seven highly abundant proteins from 200 human plasma samples. A conventional LC system was used to automate the sample injection, as well as the introduction of the two buffers that the immunodepletion process requires. The figure shows reproducible results for over 200 injections. In addition, one-dimensional gel patterns of the flow-through fractions from multiple runs (data not shown) indicated consistency and robustness of the method.


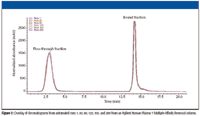
Figure 1
New Methods for Protein-Level Fractionation
To achieve the greatest number of protein identifications with LC–MS analyses, samples typically must be fractionated in multiple dimensions. Fractionation can occur either before or after protein digestion. At the peptide level, researchers often successfully reduce sample complexity via a combination of strong cation exchange and reversed-phase LC. Fractionation can occur instead (or in addition) at the protein level. Macroporous reversed-phase C18 (mRP-C18, Agilent Technologies) columns constitute one relatively new way to achieve reproducible, high-resolution separations of proteins with high recoveries.
OFFGEL electrophoresis (pI-based fractionation system developed jointly by Agilent and DiagnoSwiss S.A., Monthey, Switzerland) is another new technique for protein fractionation (1,2). It separates either proteins or peptides by isoelectric point (pI), with sample recovery in the solution phase. The solution-phase, pI-based fractionation system has the advantage of providing the protein or peptide pI, which scientists can then use to validate the results of protein database searches that are based upon MS-MS data. Both macroporous reversed-phase C18 columns and the solution-phase, pI-based fractionation system allow one to load sufficient sample so that low-level proteins can be detected in subsequent LC–MS analyses. These techniques are discussed in detail below.
Columns for High Protein Recoveries
The use of macroporous reversed-phase C18 columns is a relatively new method for protein fractionation. These columns exhibit excellent resolution, loadability, and reproducibility. Conventional reversed-phase LC columns often lack the resolution needed for complex samples like cell lysates, and protein recoveries are typically only 30–80%. The macroporous columns provide much better resolution. They also dramatically improve recoveries because a proprietary surface treatment, in combination with the large-pore bonded-silica material, prevents irreversible protein adsorption. Under optimized conditions, protein recoveries are typically greater than 95% and have been measured at 98% for immunodepleted serum (3). High recoveries eliminate sample cross-contamination and allow direct sample comparison, as shown in Figure 2.

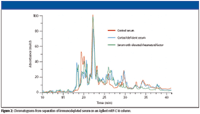
Figure 2
In addition, macroporous reversed-phase columns are ideal for desalting and concentrating protein samples (4). The process is faster than with spin concentrators and avoids the molecular weight cutoff that typically is associated with that technique.
Immunodepletion in Combination with Macroporous Reversed-Phase C18 Fractionation
For biomarker discovery in human plasma, it is very powerful to combine multiple affinity removal of highly abundant proteins (discussed earlier) with macroporous reversed-phase fractionation of the remaining proteins. The transition between the two techniques is very easy; after denaturation with urea, researchers apply the immunodepleted plasma directly onto the macroporous reversed-phase C18 column (5).
A recent study took advantage of the combination of immunodepletion and macroporous reversed-phase C18 fractionation (6). After affinity removal, the researchers concentrated, desalted, and fractionated the plasma with the macroporous reversed-phase C18 column at 80 °C. The optimized LC conditions provided high protein recoveries, enhanced peak resolution, and reproducible fractionation.
After macroporous reversed-phase fractionation, the researchers digested the proteins in solution with trypsin. They analyzed the peptides by LC–MS-MS using a polymeric-based microfluidic device in combination with an ion trap. The overall workflow minimized sample manipulations; for example, it was not necessary to perform dialysis, buffer exchange, or precipitation. The combination of affinity removal with high-capacity macroporous reversed-phase C18 fractionation and subsequent peptide analysis allowed the identification of hundreds of low-abundance plasma proteins.
Fractionation of Difficult Membrane Proteins
The macroporous reversed-phase C18 columns have proven extremely useful for studies of membrane proteins. Although membrane proteins are an excellent source of potential biomarkers, they have received less study because their hydrophobicity poses recovery challenges. On both gels and most reversed-phase LC columns, they resolve poorly and are difficult to recover. Standard C18 reversed-phase columns irreversibly bind hydrophobic proteins, causing shifts in retention time and irreproducible peak areas. The latest well-engineered macroporous reversed-phase columns overcome these problems, making the macroporous reversed-phase C18 columns excellent tools for biomarker discovery with these difficult samples (7).
A recent study showed the advantage of a macroporous reversed-phase column for fractionation of membrane proteins from HeLa cells (8). The workflow incorporated high-resolution fractionation steps but required a minimum of sample manipulations. The membrane proteins were solubilized and then were fractionated using an optimized gradient on a 50 mm × 4.6 mm macroporous reversed-phase C18 column. After combining, 17 fractions underwent in-solution digestion.
The peptides in the digests were fractionated with 11 salt steps on a capillary strong cation exchange column, and the resulting fractions were analyzed using the Agilent HPLC-Chip/MS system in combination with an ion trap. The MS-MS spectra were searched against the Swiss-Prot database (Swiss Institute of Bioinformatics, Geneva, Switzerland) using the Spectrum Mill MS Proteomics Workbench software (Agilent Technologies), and results were validated using conservative criteria. The result was identification of an unprecedented number of proteins. A total of 954 proteins were identified, including 470 membrane proteins, of which 337 were integral membrane proteins.
A number of factors enabled the identification of such a large number of membrane proteins in this difficult sample. The unique surface treatment of the macro-porous reversed-phase C18 column allowed very high protein recoveries. The column, in combination with the optimized gradient and elevated temperature, enabled excellent separation. Third, the high loadability of the macroporous reversed-phase C18 column, combined with the sensitivity of the peptide analysis system, allowed detection of numerous low-level peptides.
pI-Based Fractionation for Sample Recovery in the Liquid Phase
The solution-phase, pI-based fractionation system is a relatively new separation sytem that fractionates either proteins or peptides by pI. It achieves the same resolution as immobilized pH gradient isoelectric focusing (IPG IEF), but it removes the requirement for tedious post-IEF sample handling. After IEF, the IPG gel strips must be cut into sections and the peptides extracted and purified (9,10). With solution-phase, pI-based fractionation, the proteins or peptides end up in the liquid phase, which makes this technique directly compatible with LC–MS. There is no need for additional sample extraction or purification — steps that could lead to sample losses.
Figure 3 shows the principle of solution-phase, pI-based fractionation. The sample is diluted in focusing buffer and is loaded into a series of isolated wells. The well forms a liquid-tight seal against a rehydrated conventional IPG gel strip. Upon application of an electric field, the proteins or peptides are forced to migrate along the electric field lines through the liquid-IPG gel strip system until they reach the well where the pH of the gel is equal to the pI of the peptide or protein. As the electric field extends into the wells, the majority of proteins or peptides are conveniently recovered in the liquid phase, which contains no detergents, surfactants, or other species that interfere with LC–MS.


Figure 3
One very useful aspect of solution-phase, pI-based fractionation is that it can be applied twice in the same experiment — it can fractionate at both the protein level and the peptide level. Another convenient feature is that it requires a minimum of sample handling. Fractionated proteins can proceed directly to the digestion step, and fractionated peptides can proceed directly to LC–MS.
A unique advantage of solution-phase, pI-based fractionation is that researchers can adjust the separation by using IPG gels with various pI ranges. By using IPG gels with sufficient resolution (less than 0.2 pI units), it is possible to resolve isoforms of a protein (such as charged posttranslational modifications) and to recover them in individual solutions. Likewise, it is possible at the peptide level to identify amino acid mutations (11).
Comparison of Solution-Phase, pI-Based Fractionation with Strong Cation Exchange
A recent experiment that compared fractionation of peptides with either strong cation exchange or solution-phase, pI-based fractionation showed the advantage of solution-phase, pI-based fractionation for increased protein identifications. Immunodepleted human serum was digested tryptically. The resulting peptides were fractionated either into 24 fractions by solution-phase, pI-based fractionation (pH 3 to 10), or into 50 fractions by strong cation exchange. The individual fractions from both experiments were then analyzed using the Agilent HPLC-Chip/MS combined with an ion trap. The separation by solution-phase, pI-based fractionation produced twice the number of protein identifications and about three times the number of peptide identifications as the strong cation exchange technique.
Use of pI Values from Solution-Phase, pI-Based Fractionation for Validation of Database Search Results
Another advantage of solution-phase, pI-based fractionation is that the experimentally determined protein or peptide pIs can be used as an additional validation tool when reviewing results of protein database searches. When the software used for a protein database search also calculates and displays peptide pIs, it is easy to compare the calculated pIs with those from the solution-phase, pI-based separation. Researchers can use the experimentally derived pIs to rule out false positive matches, or they can validate additional matches with slightly lower scores when the pI values correspond as well. In a recent study, use of pI as an additional validation criterion led to 19–22% more peptide identifications (12).

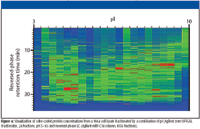
Figure 4
Very High-Resolution Fractionation for Complex Samples
For complex samples such as cell lysates, it is possible to separate proteins first by solution-phase, pI-based fractionation and second by macroporous reversed-phase LC. The transition between the two techniques is seamless, with no sample manipulation in between. The combination can provide up to 1500 individual fractions, for an extremely high-resolution separation. With a macroporous reversed-phase LC column of smaller internal diameter, it is possible to introduce the fractions directly into a time-of-flight MS for measurement of molecular mass. Alternatively, with visualization software, such as the experimental software shown in Figure 4, it is possible to compare samples and pick selected fractions for digestion and MS-MS analysis. Relative to two-dimensional gel electrophoresis, the combination of solution-phase, pI-based fractionation and macroporous reversed-phase LC provides a more information-rich, faster, more reproducible, and easier-to-use alternative.
Conclusion
Successful execution of LC–MS-based proteomic studies is highly dependent upon well-designed, seamless sample preparation schemes. For plasma and serum samples, affinity chromatography is proving to be a robust, reproducible method for removing highly abundant proteins that mask low-level proteins of interest. For many complex proteomic samples, fractionation at the protein level helps to reduce sample complexity so that it is possible to identify more proteins. While historically, researchers have used gels to fractionate proteins, new techniques such as solution-phase, pI-based fractionation, and reversed-phase separations with macroporous LC columns provide high loadability, resolution, and reproducibility, and are easier to automate. These new methods also are enabling the successful fractionation of difficult samples such as membrane proteins, as well as opening up rich possibilities for biomarker discovery.
Jerome Bailey is Program Manager, Bioseparations, Agilent Technologies, Little Falls, Delaware. Peter Mrozinski is an Application Scientist, Bioseparations, Agilent Technologies, Little Falls, Delaware. Tobias Preckel is Product Manager, OFFGEL Fractionator, Agilent Technologies, Waldbronn, Germany. Christine Miller is an Application Scientist, LC–MS, Agilent Technologies, Santa Clara, California. James Martosella is an R&D Scientist, Bioseparations, Agilent Technologies, Little Falls, Delaware. Robert Kincaid is a Senior Research Scientist, Agilent Laboratories, Santa Clara, California.
References
(1) P.E. Michel, F. Reymond, I.L. Arnaud, J. Josserand, H.H. Girault, and J.S. Rossier, Electrophoresis 24, 3–11 (2003).
(2) M. Heller, M. Ye, P.E. Michel, P. Morier, D. Stadler, M.A. Junger, R. Aebersold, F. Reymond, and J.S. Rossier, J. Proteome Res. 4(6), 2273–2282 (2005).
(3) W. Barrett, J. Martosella, N. Zolotarjova, L-S. Yang, C. Szafranski, and G. Nicol, Agilent publication number 5989-2505EN (2005).
(4) W. Barrett, J. Martosella, B. Boyes, C. Szafranski, and G. Nicol, Agilent publication number 5989-2506EN (2005).
(5) J. Martosella, N. Zolotarjova, H. Liu, G. Nicol, and B.E. Boyes, J. Proteome Res. 4(5), 1522–1537 (2005).
(6) N. Zolotarjova, J. Martosella, P. Mrozinski, H. Chen, H. Liu, W. Barrett, and J. Bailey, "Multi-Dimensional Workflow Approach for Human Plasma Analysis," Poster 231, HPLC 2006 (2006).
(7) J. Martosella, N. Zolotarjova, H. Liu, S. Moyer, P. Perkins, and B.E. Boyes, J. Proteome Res. 5(6), 1301–1312 (2006).
(8) J. Martosella, N. Zolotarjova, and H. Liu, Agilent publication number 5989-5020EN (2006).
(9) A.S. Essader, B.J. Cargile, J.L. Bundy, and J.L. Stephenson Jr., Proteomics 5, 24–34 (2005).
(10) G. Maccarrone, I. Birg, E. Malisch, M.C. Rosenhagen, C. Ditzen, J.A. Chakel, F. Mandel, A. Reimann, C.C. Doertbudak, K. Haegler, F. Holsboer, and C.W. Turck, Clinical Proteomics 1, 333–364 (2004).
(11) M. Heller, P.E. Michel, P. Morier, D. Crettaz, C. Wenz, J-D. Tissot, F. Reymond, and J.S. Rossier, Electrophoresis 26, 1174–1188 (2005).
(12) C. Miller, S.T. Hoehn, P. Hoerth, and T. Preckel, "Enhanced Reproducibility and Predictability in Serum Proteomics Achieved by Off-Gel Isoelectric Focusing of Tryptic Peptides," Poster 132, ABRF 2006 (2006).


















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


