New Frontiers for Mass Spectrometry in Lipidomics, Part II
LCGC North America
Mass spectrometry analysis of lipids in complex matrices is revolutionizing the way lipid research is conducted.
Innovative technologies are rapidly facilitating our ability to measure the numerous and diverse lipids present in biological samples. In the second and final installment of this series, we present and discuss the use of those technologies for lipid analysis.
In part I of this series, we introduced the concept of lipidomics as a new research strategy for analyzing lipids using state-of-the-art mass spectrometry (MS) technology. Together with genomics, transcriptomics, and proteomics, lipidomics is now routinely used in various scientific areas including drug and biomarker discovery, drug development, nutrition, biotechnology, and metabolic engineering research. Moreover, the future of lipidomics holds significant promise to further the development of personalized medicine.
Researchers use lipidomics as a tool to analyze a selected set of lipids (targeted lipidomics). They also use it, in a more comprehensive way, to screen all lipids present in a biological sample (untargeted lipidomics). A lipidomic research strategy presupposes the choice of analytical solutions — sample preparation, chromatography, ionization sources, and MS analyzers — that are more appropriate for answering the biological questions posed. In the second and final installment of this series, we present and discuss the use of those technologies for lipid analysis.
Sample Preparation
The high sensitivity of MS offers less labor-intensive lipid analyses. Nevertheless, the quality of sample preparation remains an important factor for success. Sample preparation includes lipid extraction from the biological matrix and elimination of nonlipid contaminants. Usually, lipid concentrations in biological samples are normalized by volume, tissue weight, cell number, or protein or DNA concentration. Quantification is done by lipids labeled with stable isotopes or nonendogenous structural analogs (internal standards) that are added before the extraction procedure. The methods for lipid extraction vary according to the sample matrix, the particular lipids under investigation, and the subsequent analytical method. The most common methods for lipid extraction are liquid–liquid extraction and solid-phase extraction (SPE).
Liquid–Liquid Extraction
Jordi Folch and colleagues (1) and E.G. Bligh and W.J. Dyer (2) developed the most widely accepted methods for lipid extraction from biological samples. Those methods are based on chloroform–methanol protocols that include phase partitioning of lipids into the organic layer. For a large variety of physiologically relevant lipids, the protocols work relatively well, and they have been adapted toward recovering more complex lipid chemistries as well as low-abundance lipid species. Multistep extractions and pH alterations also can be used for recovering acidic phospholipids such as gangliosides and phosphoinositides. Alternative extraction procedures that use less toxic organic solvents such as methyl-tert-butyl ether, hexane–isopropanol, and ethyl acetate–ethanol mixtures have been proposed for a wide range of tissues.
Solid-Phase Extraction
SPE has become a frequently used analytical procedure for rapid, preparative separation of total lipid extracts into different lipid classes. SPE involves using diverse solvents and commercial, prepacked columns with various stationary phases including silica and reversed-phase materials. (The latter use chemically bonded octadecylsilyl [ODS, C18] groups or ion-exchange media such as those with bonded aminopropyl or phenylsulfonic acid moieties.) Lipid extraction from a complex matrix might be achieved by choosing conditions (solvents and stationary phases) so that the required analyte is retained on the column while the impurities pass directly through. Conversely, extraction occurs by allowing the analyte to be eluted through the column while the impurities are retained. Targeted lipidomic studies often use SPE for fractionating the low-abundance lipids species from the highly abundant species like phospholipids, creating dynamic-range and ion-suppression issues.
Lipidomics Approaches
These three MS-based lipidomics approaches are currently in use:
- Direct infusion MS (for example, shotgun lipidomics)
- On-line chromatographic separation-MS (for example, liquid chromatography–mass spectrometry [LC–MS])
- Surface-based desorption ionization MS (for example, MS imaging)
Ion mobility spectrometry, a post-ionization separation technique, can be coupled to any of these approaches.
Direct Infusion
Directly infusing lipid extracts into the ionization source (usually electrospray ionization [ESI] or nanoESI) of the mass spectrometer without prior chromatographic separation provides for high-throughput analysis of lipid species in biological samples. This technique is known as "shotgun lipidomics" (3), by analogy to the much-used shotgun genomic sequencing. Direct infusion experiments are performed by continuous flow injection or loop injection, using automated robotic systems. "Intrasource" separation preferentially ionizes certain lipid classes by varying ESI parameter values, like source temperature and cone voltage, and doping the spray solvent with counterions, like lithium hydroxide. Additional ramping of fragmentation conditions, such as collision energy or other instrument settings, can increase the dimensions of separation of lipid species in what has been named "multidimensional MS-based shotgun lipidomics" (MDMS-SL).
Similar to proteomics experiments, shotgun lipidomics experiments lend themselves to dividing into categories of top-down and bottom-up. In a top-down analysis, lipid species are identified by their accurate mass-to-charge ratio (m/z), determined on high-resolution mass spectrometers such as time-of-flight (TOF), Fourier transform ion cyclotron resonance (FT-ICR), and orbital trap (4–6). Although individual molecular species are not identified by MS-MS, top-down lipidomics has demonstrated potential in high-throughput screens.


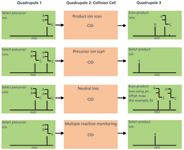
Figure 1: MS-MS operation modes for triple-quadrupole instruments. In triple-quadrupole instruments, ions pass through three successive quadrupoles (Q1, Q2, and Q3). Q2 is the collision cell, where precursor ions fragment upon collision-induced dissociation (CID) with an inert gas (helium or argon).
In bottom-up lipidomics, all lipid precursor ions are subjected to MS-MS analysis, and identified by m/z of characteristic fragments or neutral losses on triple quadrupole instruments (Figures 1 and 2) (7).


Figure 2: Representative precursor ion scanning of phospholipid classes from a total lipid extract using direct infusion-MS in positive ESI mode. [M+H]+ ions for phosphatidylcholines (PC), lyso-PC, and sphingomyelin were detected by monitoring the characteristic fragment at m/z 184.1 with a triple-quadrupole instrument.
Figure 1 shows MS-MS modes for triple-quadrupole instruments. Four main MS-MS scan experiments use a triple-quadrupole instrument:
- Product ion scan: A precursor ion is selected in Q1, fragmented in the collision cell (Q2), and then all resultant masses are scanned (Figure 1, panel A). This experiment is commonly performed to identify transitions used for quantification by tandem MS.
- Precursor ion scan: The product ion is selected in Q3, and the precursor masses are scanned in Q1 and fragmented in the collision cell (Figure 1, panel B). This experiment is performed to identify complex lipids containing common building blocks and therefore belonging to the same lipid class or subclass.
- Neutral loss scan: Q1 scans all the masses; all ions are fragmented in the collision cell. Q2 also scans, but at a set offset from the first mass analyzer (Figure 1, panel C). This offset corresponds to a neutral loss that is commonly observed for lipids loosing the same fragment on collision and which are, therefore, closely related.
- Multiple reaction monitoring (MRM): Both Q1 and Q2 are set to a selected mass (Figure 1, panel D). This experiment offers a very selective and highly sensitive analysis used mainly for quantification in targeted lipidomic approaches.
Multiple (tens) precursor-ion and neutral-loss scans could be monitored simultaneously to detect various classes of lipids for a more untargeted analysis. Indeed, most lipid species represent linear combinations of relatively few building blocks. Those building blocks include glycerol, sphingoid bases, polar head groups, and fatty acyl substituents, which can be released from the precursor ions upon collision-induced dissociation (CID) with an inert gas (argon or helium). For example, using ESI in positive mode, precursor-ion scans for m/z 184.1 (phosphocholine, Figure 3) or neutral loss scans for m/z 59.1 (trimethylamine) can identify the [M+H]+ ions that correspond to sphingomyelins, phosphatidylcholines, and lysophosphatidylcholines. Using ESI in negative mode, precursor-ion scans for m/z 97.0 (sulfate) can identify the [M-H]- ions of sulfatides whereas neutral loss scans for m/z 87.0 (serine) can be used to identify phosphatidylserines.


Figure 3: Representative data-independent analysis of a selected phosphatidylcholine species (1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine) using ESI in positive mode allows the simultaneous acquisition of precursor (top) and fragment ion (bottom) information in a single UHPLC run.
Shotgun lipidomics on triple-quadrupole instruments remains a low-mass resolution approach (1–2 Da), and only a single precursor or neutral-loss scan can be performed at a time. Hybrid, tandem mass spectrometers built on TOF, FT-ICR, and orbital trap technology now make it possible to acquire high-resolution tandem mass spectra from hundreds of lipid precursors (6,8). For example, a quadrupole-TOF instrument makes it possible to sequentially isolate precursor ions in unit or higher mass resolution windows by using a quadrupole working stepwise from a low to a high m/z ratio, transmit ions into a collision cell, and acquire accurate measurements of full product ion spectra for that window at each step of the quadrupole. The automated post-acquisition interpretation of the full product ion spectra effectively emulates an unlimited number of precursor and neutral-loss scans in a single analysis. With these modes of acquisition, no prior knowledge of the lipid ions of interest is required, which is ideal for the rapid analysis of unknown lipid mixtures in biological samples.
Although direct infusion can offer a high-throughput lipidomic technique, it is nonetheless associated with drawbacks. It cannot distinguish isobaric and isomeric species, whose masses are identical, and they often display similar fragmentation profiles. Furthermore, simultaneous introduction of multiple analytes into the ionization source can induce undesirable effects such as ion-suppression.
On-Line Chromatographic Separation-MS
LC–MS
To overcome the drawbacks of direct infusion, a more traditional approach engages the power of the LC separation to divide a complex mixture of lipids before MS analysis. LC can reduce ion suppression caused by coeluted lipids and isobaric interferences, and it can often separate isomers. In particular, chromatographic separation before an MS analysis is still necessary for analyzing very low-abundance (picomolar and attomolar levels) signaling lipids, such as eicosanoids, docosanoids endocannabinoids, and N-acyl amino acids, to name a few. Important structural information can be found in the elution order from LC columns. The elution order can significantly aid in the characterization of lipids, generating an orthogonal measure (retention time, factor, or index) that gives information on the physical-chemical properties of the lipid species analyzed. Furthermore, because lipids can differ in mass by only two units, a partial chromatographic separation helps avoid the isotopic effects, which affect the actual mass abundance during direct-infusion experiments.
The combinatorial nature of complex lipids, however, sometimes makes separating isomeric species by conventional high performance liquid chromatography (HPLC) difficult. One approach intended to increase chromatographic resolution uses reduced particle sizes (<2 µm). That approach led to the development of ultrahigh-pressure liquid chromatography (UHPLC) (10). Sub-2-µm particles provide narrow chromatographic peaks (often <3 s), which results not only in better resolution but also in lower detection limits and shorter times for the chromatographic run compared with conventional HPLC (9,10). The high pressures (10,000–15,000 psi) needed to operate these columns, which require special instruments, constitute the downside of this approach. UHPLC is often coupled to hybrid-tandem MS, which allows data-independent acquisition (see part I of this series) for the simultaneous collection of both precursor and fragment ion information (Figure 3) in a single chromatographic run (9,10).
Reversed-Phase LC
Reversed-phase LC using C18 columns is often used to separate lipids according to their fatty acyl chain length and degree of unsaturation. Lipids species containing longer acyl chains are eluted from the LC column later than shorter chain lipids (for example, C18:0>C16:0>C14:0). Saturated acyl structures tend to be eluted later than long polyunsaturated structures do (for example, C18:0>C18:1>C18:2). Generally, the first double bond on an acyl chain reduces the effective chain length by a little less than two carbon units, and additional double bonds have smaller effects on retention (9,10).
Hydrophilic Interaction Chromatography
Although reversed-phase separation methods provide good separation of lipids, the complete separation of lipid classes is not achievable. Normal-phase methods typically allow separation and characterization of different lipid classes, but their lengthy elution and equilibration times make them time-consuming processes. Moreover, the mobile phases typically used for normal-phase separations, such as chloroform or hexane, are difficult to handle because of their volatility and toxicity, and they prove challenging for ionization and introduction into a mass spectrometer. Although hydrophilic interaction liquid chromatography (HILIC) is considered a variant of normal-phase chromatography, using a reversed-phase solvent system (organic-aqueous) can avoid these limitations. Indeed, because of the high percentage of organic mobile phase used in HILIC (for example, >80% acetonitrile), ESI may be improved through more efficient mobile-phase desolvation and compound ionization (11).
Multidimensional Chromatography–MS
The ability to perform two-dimensional LC in one injection can improve the separation of complex lipid mixtures. For example, by coupling HILIC with reversed-phase chromatography, it is possible to profile different lipid classes in the first dimension (HILIC) and then separate lipid species in the second dimension (reversed phase), with significantly increased sensitivity (12). With the advent of commercialized two-dimensional UHPLC systems that couple with MS instruments, multidimensional chromatography will increasingly become a more practical tool for deconvoluting the chemical complexity of lipidomic profiles.
Supercritical Fluid Chromatography–MS
Recent advances in technology have revived the exploration of supercritical fluid chromatography (SFC) as a viable analytical technique for hydrophobic molecules (13). Supercritical fluids are highly compressed gases, which share the physical properties of both gas and liquid, and exhibit low viscosity and high solute diffusivity. These properties promote lower back pressure, shorter re-equilibration times, and higher throughput. Supercritical fluids (for example, carbon dioxide) in association with a cosolvent (typically, methanol) are compatible with MS. Moreover, they have demonstrated their usefulness as tools for separating complex mixtures of lipids. Although still in its infancy, SFC–MS can potentially find various applications for analyzing complex lipid mixtures while reducing the use of toxic solvents and the cost of analyses.
Microfluidic LC–MS
Progress in microfluidic LC has led to the integration of most of the nanoflow LC components into a single LC-chip, which avoids problems associated with capillary connections and the need to keep the system free of leaks, blockages, and excessive dead volume. Lipidomics researchers are now benefiting from the higher sensitivity of the integrated LC-chip–MS platforms when analyzing low-abundance lipids and infinitesimally small amounts of biological materials such as dried blood, skin, hair, saliva, tears, and sweat — all of which may be used for diagnostic purposes.
Surface-Based Desorption Ionization MS
The past decade has witnessed an exponential growth in surface-based desorption ionization (DI) techniques and their applications in lipid analysis (14). In DI, the ionization process begins by irradiating a defined spot on the solid-state sample using a focused, excitatory beam such as a laser, ions, or charged solvent droplets. Upon impact, the sample's surface releases a vapor of ionized molecules that can be directed into a mass spectrometer. Alternatively, acoustic or thermal desorption could initiate the ionization process.
Notably, DI-MS spectra of the biological tissues feature ions corresponding mainly to lipids. Lipids, which by molar quantities are the most abundant ionic molecular species in tissues, ionize well under physiological conditions. As occurred with the first MS experiments in the 1950s (please see part I of this series), lipids are again proving useful as a molecular tool for evaluating the potential of new MS-based technologies.
Except for matrix-assisted laser desorption ionization (MALDI), most DI techniques are matrix-free. While some operate mostly under vacuum, like MALDI, cluster-secondary ion MS (SIMS), and nanostructure-initiator mass spectrometry (NIMS), others can operate at atmospheric pressure (ambient MS techniques). There are more than 25 DI techniques currently being used, and they display unique advantages according to sample type and application (14). Generally, DI-MS provides a new level of description beyond the pure measure of lipid concentration, which is the detailed spatial distribution of lipid species on a surface. Information like this is often missed using traditional sample preparation and lipid extraction protocols for lipidomic analysis.
Nevertheless, surface-based, DI-MS techniques are not immune to drawbacks such as the incapacity to separate isobaric and isomeric species and the limitation of being (frequently) nonquantitative by nature. Apart from those drawbacks, DI-MS approaches main advantage is the ability to generate lipid fingerprints for discriminating among phenotypes, for example, healthy from diseased and bacterium A from bacterium B. Therefore, for their practicality, DI-MS techniques offer a convenient solution for the development of devices for lipidomics-based diagnostics.
Three main biological applications for DI are of interest for lipids analysis:
- DI-MS techniques are useful for real-time, rapid, in situ screening of cells, biological fluids, and tissues such as leaves, bacteria, skin, biopsies, and dried blood spots. Many new DI technologies have been applied to this kind of lipid analysis, including direct-analysis in real time (DART), extraction electrospray ionization (EESI), probe electrospray ionization (PESI), and rapid evaporative ionization mass spectrometry (REIMS). Using these techniques, the in situ generation of a particular profile of lipid ions has been proposed for the real-time molecular differentiation of diseased versus healthy tissues.
- DI-MS can be used for direct analysis of processed tissue specimens or of planar chromatography spots, for example, paper chromatography, thin-layer chromatography (TLC), and high performance TLC (HPTLC). For example, we can now analyze by DI-MS processed tissues such as lipid extracts with or without using special surfaces that favor the ionization process. Robotic systems for in situ lipid extraction (by either solvent or SPE) and deposition have been shown to increase the dynamic range of detection (15). Using DI-MS in conjunction with planar chromatography is another very convenient method to analyze lipid species. In the past, unknown lipids were scraped from the paper or TLC–HPTLC plate, eluted into a tube, and transferred into the mass spectrometer. Today, very convenient planar chromatography can be interfaced with DI-MS to automatically analyze the lipids on the paper or plates.
- DI-MS can be used for scanning through tissues and cells along the x-, and y-axes to create topographic maps of lipid composition. For that reason, we call this technique MS imaging (MSI) (16). For example, we know little about the precise distribution of lipid species in distinct brain regions, such as the hippocampus and cortex. Nor do we know much about the distribution of many lipid species in the various cellular organelles, such as nuclei and mitochondria. Laser-capture microdissection and differential centrifugation have been used to isolate fine structures of tissues and subcellular organelles. Yet such techniques generally involve processing and extracting the samples before MS analysis and may, therefore, chemically alter the lipids under study. MSI aims to detect lipids in their natural environment, by scanning through tissues and generating 2D or 3D topographical maps of lipid distribution.
MSI generates spectacular images of molecular composition of tissues highlighting the potential of DI technologies for lipid analysis (17–21) (Figure 4). Some of the first DI approaches used to map lipids from biological samples were SIMS, MALDI, and desorption electrospray ionization (DESI). In terms of spatial resolution, SIMS, MALDI, and DESI are capable of resolving features as small as 1, 20, and 200 µm, respectively. The micrometer size spatial resolution of SIMS has been used for single cell-imaging and allowed to determine the fine structure of plasma membrane domains during membrane fusion (17).


Figure 4: Imaging spleen tissue using NIMS. This image is courtesy of Dr. Gary Siuzdak (20).
SIMS suffers, however, from the fragmentation of lipids generated by the use of high-energy ion beams. Nevertheless, using polyatomic ionic particles, such as C60+ clusters, seemingly overcomes this problem (18). In MALDI, the matrix can add significant background at <1000 Da, complicating analysis of the low-mass range lipids and the spatial resolution achievable in tissue imaging is limited by the size of the matrix crystals (19). In DESI, the spatial resolution is limited by the cone of the charged solvent spray.
Because of these limitations, several new, matrix-free, DI techniques have recently been used for MS imaging of lipids. These include the NIMS (20) (Figure 4) and laser ablation ESI (LAESI) techniques (21), which can claim the advantages of low chemical noise and high sensitivity in the low-mass range as well as improved spatial resolution (Figure 4). With the development of DI technology that accommodates greater spatial resolution, the numbers of molecules to be detected becomes progressively lower, and greater sensitivity is required to analyze them. Therefore, MSI will benefit from the improvement of both DI technologies and more sensitive MS instruments.
Ion Mobility MS: A Post-Ionization Separation Tool
Ion mobility (IM) is an emerging technology that has only recently been applied to lipid analysis (22,23). Its name refers to the motion of gas-phase ions in the presence of gas (nitrogen, for instance) within a pressurized chamber (between 1 and 760 Torr), which is governed principally by the collision frequency between the ion and the gas. IM can separate gas-phase ions according to their charge and physical size (shape). IM separations occur on a time scale of milliseconds, yielding a peak capacity production rate for IM-MS of 106 to 108 s-1 , which makes it suitable for coupling with MS. IM disperses mass signals across mobility time, an additional dimension of information. IM-MS therefore benefits from an enhanced peak capacity and signal-to-noise ratio, as compared with a conventional, single dimension of MS analysis.


Figure 5: UHPLCâIM analysis of complex lipid mixtures extracted from biological samples. Abbreviations: PI, phosphatidylinositols; PC, phosphatidylcholines; PG, phosphatidylglycerols; PE, phosphatidylethanolamines; SM, sphingomyelins; DG, diacylglycerols; CE, cholesterol esters; and TG, triacylglycerols.
One primary advantage of IM-MS for lipidomic analyses lies in its ability to differentiate lipids that are isobaric in mass, but differ in structure. Lipid ions with different degrees of unsaturation, types of linkage, and polar headgroups migrate with characteristic mobility times because they differ in size (structure). Additionally, IM-MS can be combined with many sorts of lipidomic approaches, including direct infusion, like ESI-IM-MS; on-line separation, like LC-IM-MS (Figure 5); or surface DI, like DI-IM-MS (Figure 6). Because of the complexity of the biological lipidomes, the addition of mobility times as an orthogonal measurement to mass, retention times, and MS-MS fragmentation provides complementary information regarding the analyte, which adds further specificity to the lipid identification and data interpretation.

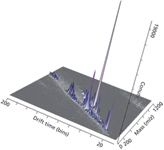
Figure 6: Representative direct analysis of human sebum in real time by diect analysis in real timeâion mobility (DART-IM).Samples were swiped on a capillary and placed near the mass spectrometer ion source and separated by ion mobility MS.
Challenges
Quantification
A true reference standard is unavailable for each target lipid species, nor is an appropriate internal standard, such as a stable isotope-labeled molecule available. Therefore, we must compromise by quantitating lipid species in biological samples. To achieve this, we normalize the individual molecular ion peak intensity using an internal standard for each lipid class. A mixture of nonendogenous molecules used as internal standards normalizes lipid levels for both extraction efficiency and instrument response. The results can be sufficiently accurate to reveal answers to biochemical questions. For instance, when comparing samples from parallel lipidomics measurements in which the precision is very high, the lower accuracy in absolute quantity is not particularly relevant.
Dynamic Range
The dynamic range in which lipid concentrations can vary in tissue may be 1012 or more (from millimolar of triglycerides to attomolar of eicosanoids). This range challenges the capability of any single approach of analysis.
Isomeric Species
The dissociation mechanisms of ionized lipids upon CID are as yet insufficient to generate a characteristic pattern of fragments that absolutely identifies some complex lipid without resorting to complicated approaches that require enzymatic or chemical reactions. For example, ambiguities often arise from regioisomerism or stereoisomerism (sn-substitution for phospholipids and triacylglycerols or double-bond position for any fatty acyl substituent). Among other examples is the difficulty of differentiating by MS lipids of identical molecular weight such as phosphatidylglucose and phosphatidylinositol or phosphatidylglycerol and lysobisphosphatidic acid.
Discovery of Novel Lipids
Although many on-line databases contain more than 20,000 lipid structures, most are based on in silico combinatorial calculations. The majority of these lipids are indeed present at very low concentration — so low as to be undetectable — or they are not actually present in real biological samples because no biosynthetic pathway exists for their formation. Furthermore, each living organism has its own unique lipid signature. Using current technology, we are able to identify less than 1000 lipids in human samples, suggesting that many novel lipids remain to be discovered.
Bioinformatics and Data Analysis
Identifying the lipid alterations associated with perturbations of biological systems or specific phenotypes is a time-limiting step of the untargeted lipidomic workflow. As such, it requires analysis of multidimensional data. With MS-based lipidomics, data sets typically comprise thousands of peaks. Each peak is associated with a mass, intensity, and additional orthogonal measurements, such as retention time (index or factor), MS-MS fragmentation data, and mobility time. Moreover, each biological sample is associated with a variety of variables, like gender, diseases, age, and treatment.
Several multivariate analysis and automatic recognition tools have managed to deconvolute the complexity of lipidomic data sets. The most commonly used solutions, however, exclude important data dimensions and therefore limit interpretation of global data sets. The incorporation of various orthogonal parameters from large sets of data in automatic recognition software could facilitate lipid identification and data interpretation.
Although, much has been borrowed from previous genomics and proteomics research, integrated solutions for lipidomics are still in their infancy. We still lack software to automate the identification of lipid species and related pathway analysis. Such a tool could be essential for interpreting data. Ultimately, new bioinformatics tools would allow integrating lipidomics data with genomics, transcriptomics, and proteomics, thus providing a systems biology approach, but at the moment the work is still in progress. Nevertheless, some on-line tools are already available including the KEGG pathway database (www.genome.jp/kegg), PubChem (pubchem.ncbi.nlm.nih.gov), and LIPID MAPS database (www.lipidmaps.org).
Conclusions
Lipidomics, a discipline that could not exist 10 years ago because of technological limitations, is now widely embraced by the scientific community. Moreover, it is revolutionizing the way we conduct research and learn about lipid biology. Lipidomic technologies appear to be fundamental to providing the correct level of description to complex biological systems. These new tools expand our knowledge about health and disease. In doing so, they force upon us a more in-depth understanding of the basic biology of living organisms, whether they be animals, plants, or microorganisms.
And yet, lipidomics still has many limitations. The implementing of comprehensive and fully automated lipidomic approaches still faces many challenges ahead. As is the case for all modern science, the development of lipidomics will benefit from an interdisciplinary effort in which a collective team of scientists of various disciplines, such as analytical chemistry, biochemistry, medicine, bioinformatics, engineering, and physics, will collaborate to ask the right questions and build the next generation of technological solutions to provide the right answers.
We can anticipate that refinements in current technologies and the addition of new orthogonal measurements will increase the sensitivity and specificity of MS analysis for lipids in complex matrices. Furthermore, considering the speed at which both technology and informatics advance, it is reasonable to hypothesize that within the next 10 years we will develop a completely new set of MS tools for lipidomic analysis that will pave the way for many scientific discoveries to come.
Acknowledgments
The author would like to thank Dave Sarro for providing helpful advice and editorial support.
Giuseppe Astarita is a principal scientist in lipidomics and metabolomics at Waters Corporation (Milford, Massachusetts) and serves as an adjunct professor at Georgetown University (Washington, D.C.). An expert in lipid MS and lipidomics for translational medicine and biomarker discovery, his work appears in several peer-reviewed scientific publications and book chapters. The International Society for the Study of Fatty Acids and Lipids has bestowed upon Dr. Astarita the Top New Investigator Award in Biochemistry of Lipids. Direct correspondence to: giuseppe_astarita@waters.com.

Giuseppe Astarita
Kate Yu "MS — The Practical Art" Editor Kate Yu joined Waters in Milford, Massachusetts, in 1998. She has a wealth of experience in applying LC–MS technologies to various application fields such as metabolite identification, metabolomics, quantitative bioanalysis, natural products, and environmental applications. Direct correspondence about this column to lcgcedit@lcgcmag.com


Kate Yu
References
(1) J. Folch et al., J. Biol. Chem.226(1), 497–509 (1957).
(2) E.G. Bligh and W.J. Dyer, Can. J. Biochem. Physiol. 37(8), 911–917 (1959).
(3) X. Han and R.W. Gross, Mass Spectrom. Rev. 24(3), 367–412 (2005).
(4) S. C. Ejsing et al., Proc. Natl. Acad. Sci. USA. 2009 106(7), 2136–2141 (2009).
(5) D. Schwudke et al., Cold Spring Harb. Perspect. Biol. 3(9), a004614 (2011).
(6) K. Schuhmann et al., Anal. Chem. 83(14), 5480–5487 (2011).
(7) B. Brugger et al., Proc. Natl. Acad. Sci. USA. 94, 2339–2344 (1997).
(8) M. Ståhlman et al., J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877(26), 2664–2672 (2009).
(9) P.D. Rainville et al., J. Proteome Res. 6(2), 552–558 (2007).
(10) J.M. Castro-Perez et al., J. Proteome Res. 9, 2377–2389 (2010).
(11) L. Zheng et al., Rapid Commun. Mass Spectrom. 24(14), 2074–2082 (2010).
(12) H. Nie et al., J. Lipid Res. 51(9), 2833–2844 (2010).
(13) T. Bamba et al., J. Biosci. Bioeng. 105(5), 460–469 (2008).
(14) G.A. Harris et al., Anal. Chem. 83(12), 4508–4538 (2011).
(15) V. Kertesz and G.J. Van Berkel, J. Mass Spectrom. 45, 252–260 (2010).
(16) J.C. Vickerman, Analyst 136(11), 2199–2217 (2011).
(17) S.G. Ostrowski et al., Science 305, 71–73 (2004).
(18) C.A. Barnes et al., Anal. Chem. 84(2), 893–900 (2012).
(19) K.A. Berry et al., Chem. Rev. 111(10), 6491–6512 (2011).
(20) M.P. Greving et al., Anal. Chem. 83(1), 2–7 (2011).
(21) P. Nemes and A. Vertes, J. Vis. Exp. 43, 2097 (2010).
(22) A.A. Shvartsburg et al., J. Am. Soc. Mass Spectrom. 22(7), 1146–1155 (2011).
(23) M. Kliman et al., Biochim. Biophys. Acta. 1811(11), 935–945 (2011).















Evaluating Body Odor Sampling Phases Prior to Analysis
April 23rd 2025Researchers leveraged the advantages of thermodesorption, followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC×GC/TOF-MS), to compare and assess a variety of sampling phases for body odor.



