Modifying the Metal Surfaces in HPLC Systems and Columns to Prevent Analyte Adsorption and Other Deleterious Effects
Interactions of certain analytes with metal surfaces in high performance liquid chromatography (HPLC) instruments and columns cause a range of deleterious effects, including peak broadening and tailing, low peak areas, and the formation of new peaks due to chemical reactions. To mitigate these effects, we have developed a novel surface modification technology in which a hybrid organic/inorganic surface based on an ethylene-bridged siloxane chemistry is applied to the metal components in HPLC instruments and columns. We demonstrate the impact of this technology on peak symmetry, peak area, and injection-to-injection and column-to-column reproducibility for several metal-sensitive analytes. We also show an example of the mitigation of an on-column oxidation reaction. A variant of this technology has recently been developed for size-exclusion chromatography of proteins. An example is shown demonstrating the use of this variant applied to size-exclusion columns for the separation of a monoclonal antibody monomer and higher molecular weight species. Together, these results highlight the importance of preventing interactions of analytes with metal surfaces in HPLC in order to achieve accurate and precise results.
The need for high pressure tolerance in high performance liquid chromatography (HPLC) instruments and columns has led to the widespread use of metal components, most commonly made from stainless steel. However, some analytes interact with the oxide layer present on the metal surfaces in the flow path of HPLC systems, resulting in a range of deleterious effects from peak broadening and tailing to reduced peak areas and high carryover (1–13). Molecules that show these effects typically contain phosphate or carboxylate groups. Examples include oligonucleotides, acidic and phosphorylated peptides and proteins, acidic carbohydrates, certain phospholipids, and small molecules including organophosphates and carboxylic acids.
In addition to these problems, conventional HPLC systems and columns may release metal ions into the mobile phase (3,14,15). These metal ions may form chelation complexes with analytes that have Lewis base groups, such as amino, hydroxyl, and carbonyl groups (1). This may be problematic when using mass spectrometry (MS) detection because of the formation of metal ion adducts which complicate mass spectra (5,16,17). It can also be an issue for analytes that undergo reactions catalyzed by metal ions. For example, amines have been shown to be prone to nitrosation when separated using conventional stainless steel systems and columns with mobile phases containing acetonitrile and ammonium hydroxide (18). Peptides and proteins are also susceptible to metal ion-catalyzed degradation during separations, as methionine residues are prone to oxidation (19). Epimerization (3) and dimerization (20) have also been reported to occur for small molecule pharmaceuticals, due to Fe3+ released from stainless steel components and adsorbed on the stationary phase in reversed-phase columns.
One approach to mitigate these problems is to use alternative metals in place of stainless steel for columns and components in the flow path of HPLC systems (21). Examples include titanium and nickel-cobalt alloys (for example, MP35N). While these alternatives exhibit improved corrosion resistance, making them useful for applications that require mobile phases with high salt concentrations, they still adsorb acidic analytes. The surface oxide layer of MP35N contains chromium oxide (22), similar to that of stainless steel. The titanium oxide layer present on the surface of titanium strongly adsorbs analytes containing phosphate groups, as shown by a <5% recovery of adenosine triphosphate measured for untreated titanium frits (16,23). Titanium is also susceptible to corrosion when exposed to anhydrous methanol, forming Ti4+ ions that may adsorb on stationary phases, causing issues that are similar to those resulting from Fe3+ released from stainless steel (3,24).
Polyether ether ketone (PEEK) is another material that has been used in place of stainless steel in HPLC columns and systems. While this engineering plastic has been used with pressures below about 5000 psi, it lacks the mechanical strength to tolerate higher pressures unless cladded with steel. In addition, PEEK is not recommended for use with some solvents, notably tetrahydrofuran, dimethyl sulfoxide, chloroform, and methylene chloride. PEEK is also relatively hydrophobic, and may cause analyte adsorption due to hydrophobic interactions (25,26). The internal diameter of PEEK tubing is also more variable than that of metal tubing, which leads to greater column-to-column retention time variability for PEEK-lined steel columns (27).
An alternative approach to mitigating these interactions is to add reagents to the mobile phase or the sample that block the binding sites. Examples include phosphate buffers and chelators, such as ethyl-enediaminetetraacetic acid (EDTA), citrate, acetylacetone, and medronic acid (1,5,7,10,28–31). However, phosphate buffers and EDTA are not volatile, making them incompatible with mass spectrometry detection. Even when using volatile additives with MS detection, ion suppression may occur (30,31). In addition, long equilibration times are often associated with the use of these types of additives (32). It is also known that chelators such as EDTA and citric acid increase the rate of corrosion of stainless steel (14).
In 2020, a new solution to the problems caused by metal surfaces was introduced, obviating the need for chelators in the mobile phase (33). Using a proprietary process, the metal components of the HPLC flow path and column were treated to form a hybrid organic/inorganic barrier covering the oxide layer on the metal surfaces (16). This process was termed hybrid surface technology (HST). A composition that is similar to that of the particles used in ethylene-bridged hybrid inorganic–organic stationary phases (34) was shown to be stable over a wide pH range (from 1–12) (33). This composition is well-suited for reversed-phase chromatography, as demonstrated for separations of oligonucleotides (16,23,35–37), acidic and phosphorylated pep- tides (38,39), certain small molecule pharmaceuticals (23,40,41), veterinary drugs (42), vitamins (43), and phospholipids (44). This composition has also been shown to be beneficial for mixed-mode reversed-phase/anion-exchange separations of tricarboxylic acid cycle metabolites (17) and acidic glycans (45), as well as hydrophilic-interaction chromatography (HILIC) separations of nucleotides and other cellular metabolites (46–48). Most recently, a variant of this composition termed hydrophilic hybrid surface technology (h-HST) was introduced for use in size-exclusion and ion-exchange columns for separations of proteins and nucleic acids (49). Here, we demonstrate the performance of HPLC systems and columns that incorporate these technologies for analytes ranging from small molecules to proteins.
Materials and Methods
Conventional ultrahigh-pressure liquid chromatography (UHPLC) instruments and columns were compared to versions employing hybrid surface technology (HST), designated as Acquity Premier Systems and MaxPeak Premier Columns, respectively (Waters).
Comparison of HST vs. Conventional Systems and Columns for a Nucleotide Separation
Separations of adenosine monophosphate (AMP) and adenosine triphosphate (ATP) were carried out using both conventional and HST Acquity UPLC Systems with Acquity UPLC BEH Amide 1.7 μm particles packed into either conventional stainless steel or HST column hardware, both 2.1 x 50 mm. The mobile phase was 65:35 v/v acetonitrile:aqueous 60 mM pH 6.8 ammonium acetate and the flow rate was 0.5 mL/min. The sample contained 20 μg/mL each of adenosine monophosphate disodium salt and adenosine triphosphate disodium salt hydrate dissolved in 95:5 v/v acetonitrile:water and the injection volume was 1.0 μL. The columns were maintained at 30 °C and ultraviolet (UV) detection was used (λ = 260 nm).
Comparison of HST vs. Conventional Columns for the Separation of a Mixture of Twelve Nucleotides
Separations of 12 nucleotides were carried out using an Acquity Premier BSM System with Atlantis BEH Z-HILIC 1.7 μm particles packed into either conventional stainless steel or HST column hardware, both 2.1 x 150 mm. The sample contained 50 μg/mL each of AMP disodium salt, adenosine diphosphate (ADP) disodium salt hydrate, ATP disodium salt hydrate, cytidine monophosphate (CMP) disodium salt, cytidine diphosphate (CDP) sodium salt hydrate, cytidine triphosphate (CTP) disodium salt, guanosine monophosphate (GMP) disodium salt hydrate, guanosine diphosphate (GDP) sodium salt, guanosine triphosphate (GTP) sodium salt hydrate, uridine monophosphate (UMP) disodium salt, uridine diphosphate (UDP) disodium salt hydrate, and uridine triphosphate (UTP) trisodium salt hydrate dissolved in 95:5 v/v acetonitrile/water. The injection volume was 1.0 μL. The mobile phase was 70:5:25 v/v/v acetonitrile/methanol/40 mM pH 9.0 ammonium bicarbonate and the flow rate was 0.15 mL/min. The separation was carried out at ambient temperature (27 °C) with UV detection (λ = 260 nm).
Determination of Peak Area vs. Mass Load for HST and Conventional Systems and Columns
Experiments were carried out using both conventional and HST Acquity UPLC BSM Systems with PDA detectors and Acquity UPLC BEH C18 1.7 μm particles packed into conventional stainless steel or HST column hardware, both 2.1 x 50 mm. A set of samples containing hydrocortisone sodium phosphate at concentrations ranging from 1–100 μg/mL in 5:95 acetonitrile/10 mM ammonium formate pH 3.0 (aq) were used, with a 2 μL injection volume. Triplicate injections were made for each sample, and the mean peak areas (λ = 246 nm) were calculated. A linear acetonitrile gradient from 5–95% in 5.3 min was employed with an aqueous mobile phase containing 10 mM pH 3.0 ammonium formate. The flow rate was 0.5 mL/min.
Comparison of Column-to-Column and Injection-to-Injection Reproducibility for HST and Conventional Columns
Experiments were carried out using a prototype Acquity Premier Quaternary System with an Acquity QDa Mass Detector. The same batch of Atlantis BEH C18 AX 1.7 μm particles was packed into six conventional stainless steel columns and six HST columns (all 2.1 x 50 mm). The sample was a 100 μg/mL aqueous solution of fructose 1,6-bisphosphate trisodium salt hydrate. Ten consecutive 2 μL injections were made on each column using an aqueous solution of 10 mM pH 3.0 ammonium formate as the mobile phase at a flow rate of 0.5 mL/min. The peaks were detected using negative mode electrospray ionization with selected ion recording at m/z = 339. The samples were maintained at 10 °C, and the columns at 30 °C.
Investigation of On-Column Oxidation for HST and Conventional Columns
Separations were carried out using a conventional Acquity UPLC I-Class System with a TUV detector, with the analyte detected at a wavelength of 290 nm. Conventional stainless steel and HST 2.1 x 50 mm columns packed with Acquity UPLC BEH C18 1.7 μm particles were used with a linear 25–80% acetonitrile gradient in 10.31 min employing an aqueous mobile phase containing 0.05% (w/v) ammonium hydroxide. The sample contained 6 mg/mL clozapine in 20:80:0.08 acetonitrile:water:acetic acid, and the injection volume was 0.25 μL. The sample was maintained at 4 °C, and the columns at 30 °C.
Comparison of h-HST vs. Conventional Columns for SEC Separations of a Monoclonal Antibody
Size-exclusion separations were carried out using an Acquity UPLC H-Class Bio System with a TUV detector equipped with a 1500 nL titanium flow cell. Conventional stainless steel and h-HST 4.6 x 150 mm columns packed with XBridge Protein SEC 250 Å 2.5 μm particles were used with aqueous mobile phases containing 100 mM pH 6.8 sodium phosphate and varying concentrations of NaCl (0–200 mM). The flow rate was 0.35 mL/min. The sample contained 2 mg/mL NISTmAb (NIST reference material 8671), and the injection volume was 1 μL. The samples were maintained at 8 °C, and the columns at 30 °C.
Results and Discussion
A sample injected into an HPLC system interacts with a number of metal components between the injector and the detector. This includes the injector needle, the seat port, the preheater, connection tubing, and the column. For a typical Acquity UPLC System with a UV detector and a 2.1 x 50 mm column, it has been estimated that the column contributes 71% of the total metal surface area contacted by the sample, while the system accounts for the remaining 29% (23). The column frits alone contribute 52% of the total metal surface area. Indeed, column frits have often been identified as the cause of many of the deleterious effects associated with metal surfaces in HPLC. However, to obtain the best results, all of these surfaces need to be modified. This approach is enabled by HST, since the technology has been found to effectively treat all of the metal components mentioned above (16).
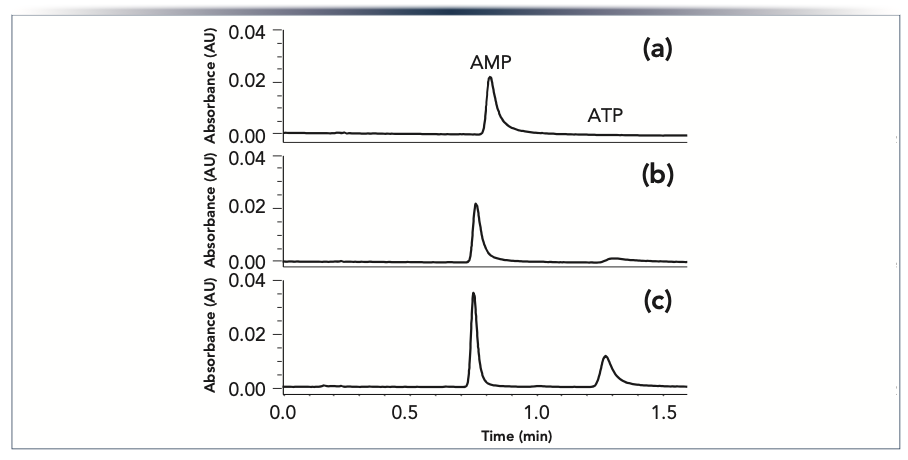
An example demonstrating the importance of modifying the surfaces in both the system and the column is shown in Figure 1. A sample containing adenosine monophosphate (AMP) and adenosine triphosphate (ATP) was separated using (a) a conventional system and conventional column; (b) a conventional system with an HST column; and (c) an HST system with an HST column. With the conventional system and conventional column, no peak was seen for ATP, which is known to adsorb strongly on metal surfaces (6,10,16,23,31,32,46,48). While a peak was observed for AMP, it showed significant tailing. When an HST column was used on the same conventional system, ATP was detected as a severely tailed peak with a low peak area. Using the HST column on an HST system, an ATP peak was observed with the expected peak area, and only slight tailing. This demonstrates the importance of modifying the surfaces in both the column and the system for analytes that exhibit strong adsorption on metal surfaces.

 a conventional UHPLC system and column; (b) the same system with an HST column; and (c) an HST system and HST column. The nucleotides were injected at 20 ng mass loads on 2.1 x 50 mm columns packed with Acquity UPLC BEH Amide 1.7 μm particles and separated using a 65:35 (v/v) acetonitrile:aqueous 60 mM pH 6.8 ammonium acetate mobile phase. The peaks were detected by absorbance at 260 nm.")
FIGURE 1: Chromatograms showing the separation of AMP and ATP obtained using (a) a conventional UHPLC system and column; (b) the same system with an HST column; and (c) an HST system and HST column. The nucleotides were injected at 20 ng mass loads on 2.1 x 50 mm columns packed with Acquity UPLC BEH Amide 1.7 μm particles and separated using a 65:35 (v/v) acetonitrile:aqueous 60 mM pH 6.8 ammonium acetate mobile phase. The peaks were detected by absorbance at 260 nm.
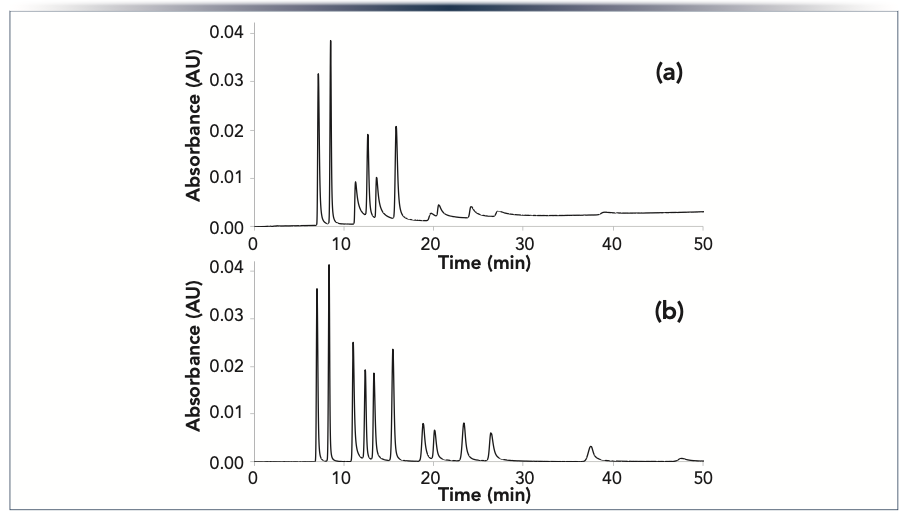
The ability to obtain narrow peaks and accurate peak areas for analytes that interact with metal surfaces is important in a number of applications, including studies of polar cellular metabolites (28,30). As demonstrated in Figure 1, nucleotides are problematic when using conventional UHPLC systems and columns. In Figure 2, we show the separation of a mixture of twelve nucleotides using an HST system with either a conventional column or an HST column. The results show dramatic improvements in peak height, peak width, and peak shape when using the HST column, particularly for the nucleotide di- and triphosphates.

 a conventional column; or (b) an HST column. The columns were 2.1 x 150 mm, and packed with 1.7 μm Atlantis BEH Z-HILIC particles. The mobile phase was 70:5:25 v/v/v acetonitrile/methanol/40 mM pH 9.0 ammonium bicarbonate, and the peaks were detected by absorbance at 260 nm. Peak identifications, left to right: AMP, UMP, ADP, CMP, UDP, GMP, ATP, CDP, UTP, GDP, CTP, and GTP.")
FIGURE 2: Chromatograms showing the separation of 12 nucleotides obtained using an HST system with (a) a conventional column; or (b) an HST column. The columns were 2.1 x 150 mm, and packed with 1.7 μm Atlantis BEH Z-HILIC particles. The mobile phase was 70:5:25 v/v/v acetonitrile/methanol/40 mM pH 9.0 ammonium bicarbonate, and the peaks were detected by absorbance at 260 nm. Peak identifications, left to right: AMP, UMP, ADP, CMP, UDP, GMP, ATP, CDP, UTP, GDP, CTP, and GTP.
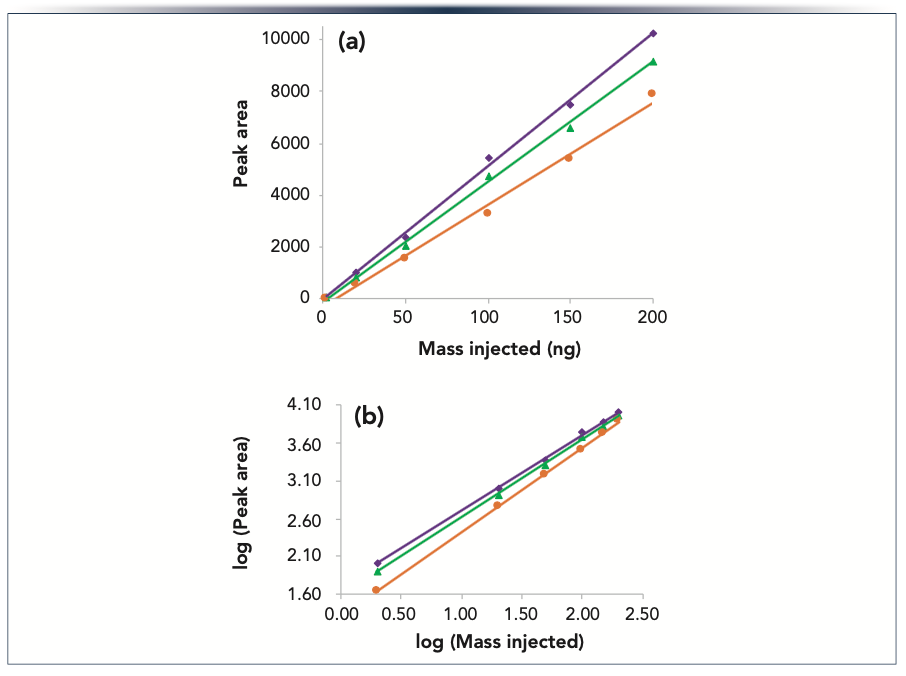
While the largest increases in peak area observed for HST versus conventional systems and columns have been reported for low analyte mass loads (16,41), significant improvements may be seen over a wide range of loads. An example is shown in Figure 3 for the steroid prodrug hydrocortisone sodium phosphate. As in our first example, we compared the results using (a) a conventional system and conventional column; (b) a conventional system with an HST column; and (c) an HST system with an HST column. The masses injected ranged from 2–200 ng. The peak areas are plotted versus mass injected in Figure 3a, and a log-log plot of the same data is shown in Figure 3b. The highest peak areas were observed using the combination of an HST system and an HST column, while the lowest were seen for the conventional system with a conventional column. The latter results also showed the lowest correlation coefficient (0.992 vs 0.998 for the other two linear correlations), indicating a greater deviation from a linear dependence of peak area on injected mass (Figure 3a). As evident in the log-log plot (Figure 3b), the relative differences in peak area were greatest for the lowest injected masses. For the 2 ng mass load, the average peak area for the HST system with an HST column was 25% greater than that for the conventional system with an HST column, and 58% greater than that for the conventional system with a conventional column.

 Plot of peak area vs. injected mass for hydrocortisone sodium phosphate obtained using a conventional system with a conventional column (orange circles), a conventional system with an HST column (green triangles), and an HST system with an HST column (purple diamonds). The lines are linear least squares fits of the data; (b) Log-log plot of the same results. Columns (2.1 x 50 mm) packed with Acquity UPLC BEH C18 1.7 μm particles were used with an acetonitrile gradient and a 10 mM pH 3.0 ammonium formate aqueous mobile phase. The peaks were detected by absorbance at 246 nm.")
FIGURE 3: (a) Plot of peak area vs. injected mass for hydrocortisone sodium phosphate obtained using a conventional system with a conventional column (orange circles), a conventional system with an HST column (green triangles), and an HST system with an HST column (purple diamonds). The lines are linear least squares fits of the data; (b) Log-log plot of the same results. Columns (2.1 x 50 mm) packed with Acquity UPLC BEH C18 1.7 μm particles were used with an acetonitrile gradient and a 10 mM pH 3.0 ammonium formate aqueous mobile phase. The peaks were detected by absorbance at 246 nm.
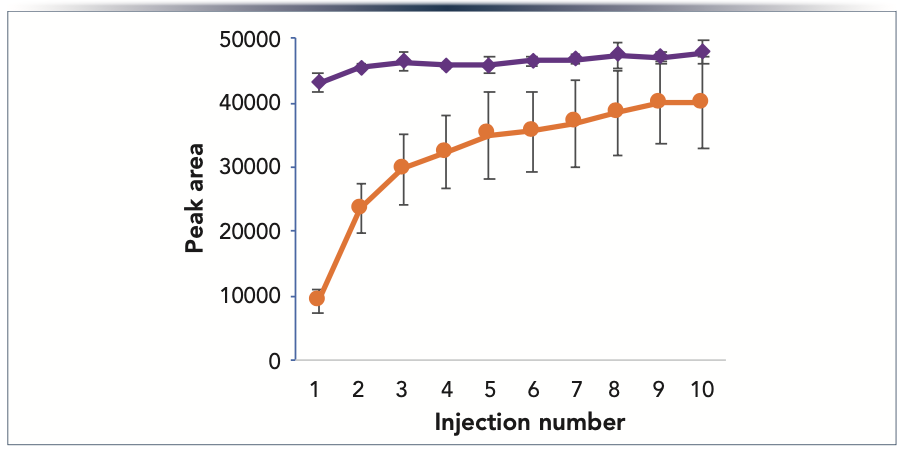
Adsorption on metal surfaces in the column hardware is a source of column-to-column variability for some analytes. This is demonstrated by the results shown in Figure 4, which compares peak areas for fructose 1,6-bisphosphate obtained using a mixed-mode reversed-phase anion-exchange stationary phase packed into either conventional or HST column hardware. For each column, a series of ten consecutive injections was made of 200 ng of fructose 1,6-bisphosphate. The error bars in Figure 4 indicate ± one standard deviation for six columns of each type. The relative standard deviations (RSD) for peak areas in the tenth injections were 3.9% for the HST columns, versus 17.7% for the conventional columns. Comparing the variability of the average peak areas across the ten injections, the RSD for the HST columns was 2.9% versus 36.5% for the conventional columns. These results show dramatic reductions in both column-to-column and injection-to-injection variability when using HST column hardware.

 and HST columns (purple diamonds) packed with 1.7 μm Atlantis BEH C18 AX particles. The mobile phase was an aqueous solution of 10 mM pH 3.0 ammonium formate. The error bars show + one standard deviation for six columns of each type. The peaks were detected by negative ion electrospray ionization mass spectrometry (m/z = 339).")
FIGURE 4: Plot of average peak area vs. injection number for fructose 1,6-bisphosphate using conventional (orange circles) and HST columns (purple diamonds) packed with 1.7 μm Atlantis BEH C18 AX particles. The mobile phase was an aqueous solution of 10 mM pH 3.0 ammonium formate. The error bars show + one standard deviation for six columns of each type. The peaks were detected by negative ion electrospray ionization mass spectrometry (m/z = 339).
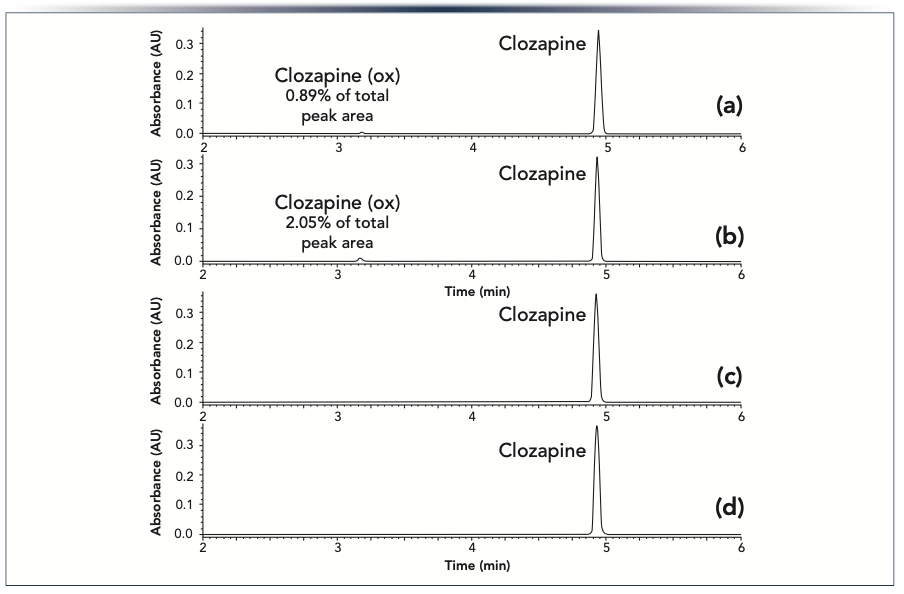
In addition to adsorption, metal surfaces or metal ions released from them have been reported to catalyze chemical reactions of certain analytes. We investigated the amount of on-column oxidation for conventional and HST columns for the pharmaceutical clozapine, using a mobile phase containing ammonium hydroxide and acetonitrile. As shown in Figure 5a and 5b, when using the conventional column a new peak appears, which has been shown by mass spectrometry to be due to an oxidized form of clozapine (50). The peak area of the oxidation product relative to the parent drug increased over a series of consecutive injections, from 0.89% in the second injection to 2.05% in the thirteenth. Similar behavior was reported by Myers and associates (18) for the on-column formation of a nitrosation impurity. It was hypothesized that the protective chromium oxide layer on the stainless steel frits is gradually lost over a series of chromatographic runs, exposing a reactive surface. At the same time, metal ions eluted from the frit may adsorb on the stationary phase and catalyze the nitrosation reaction. When using an HST column we did not observe the clozapine oxidation product (Figure 5c and 5d). This shows that HST is effective at mitigating an on-column reaction catalyzed by stainless steel columns or metal ions released from them.

 the second and (b) the thirteenth injections of clozapine on a conventional column. The clozapine oxidation product (clozapine (ox)) was formed due to an on-column reaction. Chromatograms for (c) the second and (d) the thirteenth injections of clozapine on an HST column. The columns (2.1 x 50 mm) were packed with Acquity UPLC BEH C18 1.7 μm particles. An acetonitrile gradient was used with an ammonium hydroxide aqueous mobile phase. The peaks were detected by absorbance at 290 nm.")
FIGURE 5: Chromatograms for (a) the second and (b) the thirteenth injections of clozapine on a conventional column. The clozapine oxidation product (clozapine (ox)) was formed due to an on-column reaction. Chromatograms for (c) the second and (d) the thirteenth injections of clozapine on an HST column. The columns (2.1 x 50 mm) were packed with Acquity UPLC BEH C18 1.7 μm particles. An acetonitrile gradient was used with an ammonium hydroxide aqueous mobile phase. The peaks were detected by absorbance at 290 nm.
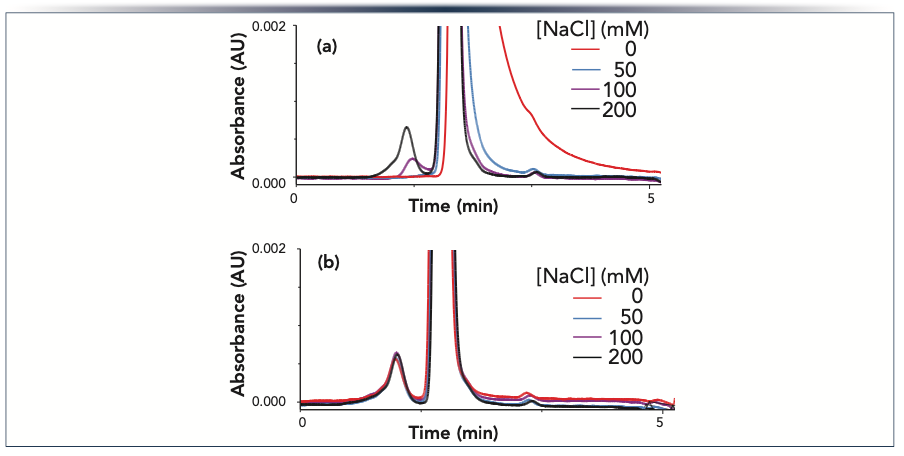
An important feature of HST is that different chemistries may be used to match the surface properties to the needs of the chromatographic mode and the intended analytes. A new variant of HST was recently developed for use in size exclusion chromatography (SEC) columns for proteins (49). This variant was termed hydrophilic HST (h-HST). An example demonstrating the benefit of this technology is shown in Figure 6. The same batch of stationary phase was packed into a conventional column and column hardware modified with h-HST. SEC separations of a monoclonal antibody monomer, fragments, and higher molecular weight species (HMWS) were carried out using mobile phases containing different concentrations of sodium chloride, which is used to mitigate ionic interactions (51). When using the conventional column, we observed a strong dependence of peak width and tailing on the salt concentration, with lower concentrations resulting in peaks that were broader and had larger tailing factors. The peak from the HMWS, which elute before the monomer, showed a greater sensitivity to the salt concentration than did the monomer peak. Only at the highest salt concentration (200 mM) was accurate quantification of the HMWS achievable. In contrast, the h-HST column showed very similar peak widths and tailing factors across the entire range of salt concentrations, as well as a consistent relative peak area for the HMWS. This is important, because it allows SEC separations to be carried out using mobile phases that are less likely to change the concentration of reversible aggregates, as has been reported to occur for high ionic strength mobile phases (52).

 a conventional column; or (b) an h-HST column, both packed with XBridge SEC 250 Å 2.5 μm particles. The mobile phase was a 100 mM aqueous solution of sodium phosphate (pH 6.8) containing varying concentrations of sodium chloride. The peaks were detected by absorbance at 280 nm. Peak identifications, left to right: high molecular weight species, monomer, and fragments.")
FIGURE 6: Chromatograms showing the separation of NISTmAb monomer, fragments, and high molecular weight species using (a) a conventional column; or (b) an h-HST column, both packed with XBridge SEC 250 Å 2.5 μm particles. The mobile phase was a 100 mM aqueous solution of sodium phosphate (pH 6.8) containing varying concentrations of sodium chloride. The peaks were detected by absorbance at 280 nm. Peak identifications, left to right: high molecular weight species, monomer, and fragments.
Conclusion
Preventing interactions of analytes with metal surfaces in HPLC systems and columns is important for obtaining accurate and precise results. Modifying the metal surfaces using hybrid surface technology has been shown to be highly effective at mitigating a range of issues, providing improved peak areas and peak shapes as well as reduced injection-to-injection and column-to-column variability. We also demonstrated mitigation of an oxidation reaction catalyzed by metal surfaces or metal ions released from them. Improvements were demonstrated for chromatographic modes including reversed-phase, HILIC and SEC. This technology has proven to be valuable for a wide range of applications, including analyses of both small and large molecule pharmaceuticals, as well as cellular metabolites, veterinary drugs and vitamins.
References
(1) S.M. Cramer, B. Nathanael, and C. Horváth, J. Chromatogr. 295, 405–411 (1984).
(2) P.C. Sadek, P.W. Carr, L.D. Bowers, and L.C. Haddad, Anal. Biochem. 144, 128–131 (1985).
(3) M.R. Euerby, C.M. Johnson, I.D. Rushin, and D.A.S.S. Tennekoon, J. Chromatogr. A 705, 229–245 (1995).
(4) A. Wakamatsu, K. Morimoto, M. Shimizu, and S. Kudoh, J. Sep. Sci. 28, 1823–1830 (2005).
(5) S. Liu, C. Zhang, J.L. Campbell, H. Zhang, K.K.-C. Yeung, V.K.M. Han, and G.A. Lajoie, Rapid Commun. Mass Spectrom. 19, 2747–2756 (2005).
(6) R. Tuytten, F. Lemière, E. Witters, W. Van Dongen, H. Slegers, R.P. Newton, H. Van Onckelen, and E.L. Esmans, J. Chromatogr. A 1104, 209–221 (2006).
(7) Y. Asakawa, N. Tokida, C. Ozawa, M. Ishiba, O. Tagaya, and N. Asakawa, J. Chromatogr. A 1198-1199, 80–86 (2008).
(8) A. Fleitz, E. Nieves, C. Madrid-Aliste, S.J. (29) Fentress, L.D. Sibley, L.M. Weiss, R.H. Angeletti, and F.-Y. Che, Anal. Chem. 85, 8566–8576 (2013).
(9) H. Sakamaki, T. Uchida, L.W. Lim, and T. Takeuchi, J. Chromatogr. A 1381, 125–131 (2015).
(10) J.C. Heaton and D.V. McCalley, J. Chromatogr. A 1427, 37–44 (2016).
(11) J.J. Hsiao, G.O. Staples, and D.R. Stoll, LCGC N. Am. 38, 146–150 (2020).
(12) H. Sakamaki, T. Uchida, L.W. Lim, and T. Takeuchi, Anal. Sci. 31, 91–97 (2015).
(13) L. Heinle, H. Patel, G. Jenkins, and K. Ruterbories, J. Chromatogr. A 1606, 460379 (2019).
(14) K.E. Collins, C.H. Collins, and C.A. Bertran, LCGC North Am. 18, 600–608 (2000).
(15) P.R. Haddad and R.C.L. Foley, J. Chromatogr. 407, 133–140 (1987).
(16) M. DeLano, T.H. Walter, M.A. Lauber, M. Gilar, M.C. Jung, J.M. Nguyen, C. Boissel, A.V. Patel, A. Bates-Harrison, and K.D. Wyndham, Anal. Chem. 93, 5773–5781 (2021).
(17) K.M. Smith, I.D. Wilson, and P.D. Rainville, Anal. Chem. 93, 1009–1015 (2021).
(18) D.P. Myers, E.M. Hetrick, Z. Liang, C.E. Hadden, S. Bandy, C.A. Kemp, T.M. Harris, and W.W. Baertschi, J. Chromatogr. A 1319, 57–64 (2013).
(19) L. Zang, T. Carlage, D. Murphy, R. Frenkel, P. Bryngelson, M. Madsen, and Y. Lyubarskaya, J. Chromatogr. B. 895-896, 71–76 (2012).
(20) Q. Wang, B.L. He, J. Zhang, Y. Huang, and B. Kleintop, J. Pharm. Biomed. Anal. 111, 288–296 (2015).
(21) S. Kromidas, Ed., The HPLC Expert II (Wiley-VCH, Berlin, Germany, 2017).
(22) A. Nagai, Y. Tsutsumi, Y. Suzuki, K. Katayama, T. Hanawa, and K. Yamashita, Appl. Surf. Sci. 258, 5490–5498 (2012).
(23) M. Gilar, M. DeLano, and F. Gritti, J. Chromatogr. A 1650, 462247 (2021).
(24) M. De Pra, G. Greco, M.P. Krajewski, M.M. Martin, E. George, N. Bartsch, and F. Steiner, J. Chromatogr. A 1611, 460619 (2020).
(25) J. Lough, M.J. Mills, and J. Maltas, J. Chromatogr. A 726, 67–75 (1996).
(26) P. Hambleton, W.J. Lough, J. Maltas, and M. Mills, J. Liq. Chromat. Rel. Tech. 18, 3205–3217 (1995).
(27) M. DeLoffi, Waters Corp. application note 720007210, 2021.
(28) K.T. Myint, T. Uehara, K. Aoshima, and Y. Oda, Anal. Chem. 81, 7766–7772 (2009).
(29) D. Winter, J. Seidler, Y. Ziv, Y. Shiloh, and W.D. Lehmann, J. Proteome Res. 8, 418–424 (2009).
(30) D. Siegel, H. Permentier, and R. Bischoff, J. Chromatogr. A 1294, 87–97 (2013).
(31) J.J. Hsiao, O.G. Potter, T.-W. Chu, and H. Yin, Anal. Chem. 90, 9457–9464 (2018).
(32) J.J. Hsiao, T.-W. Chu, O.G. Potter, G.O. Staples, and D.R. Stoll, LCGC N. Am. 38, 431–434 (2020).
(33) M. Lauber, T.H. Walter, M. Gilar, M. DeLano, C. Boissel, K. Smith, R. Birdsall, P. Rainville, J. Belanger, and K. Wyndham, Waters Corp. white paper 720006930EN, 2020.
(34) K.D. Wyndham, J.E. O’Gara, T.H. Walter, K.H. Glose, N.L. Lawrence, B.A. Alden, G.S. Izzo, C.J. Hudalla, and P.C. Iraneta, Anal. Chem. 75, 6781–6788 (2003).
(35) A. Goyon, B. Scott, K. Kurita, C.M. Crittenden, D. Shaw, A. Lin, P. Yehl, and K. Zhang, Anal. Chem. 93, 14792–14801 (2021).
(36) G.J. Guimaraes, J.M. Sutton, M. Gilar, M. Donegan, and M.G. Bartlett, J. Pharm. Biomed. Anal. 208, 114439 (2022).
(37) J.M. Nguyen, M. Gilar, B. Koshel, M. Donegan, J. MacLean, Z. Li, and M.A. Lauber, Bioanalysis 13, 1233–1244 (2021).
(38) R.E. Birdsall, J. Kellett, S. Ippoliti, N. Ranbaduge, M.A. Lauber, Y.Q. Yu, and W. Chen, J. Chromatogr. B 1179, 122700 (2021).
(39) C.J. Hughes, L.A. Gethings, I.D. Wilson, and R.S. Plumb, J. Chromatogr. A 1673, 463014 (2022).
(40) R.S. Plumb, L.A. Gethings, A. King, L.G. Mullin, G. Maker, R. Trengove, and I.D. Wilson, J. Pharm. Biomed. Anal. 200, 114076 (2021).
(41) N. Tanna, L.G. Mullin, P.D. Rainville, I.D. Wilson, and R.S. Plumb, J. Chromatogr. B 1179, 122825 (2021).
(42) K. Lo, P. Kukkadapu, P. Wagh, and M. Ritchie, Waters Corp. application note 720007545, 2022.
(43) J. Yang and P.D. Rainville, Waters Corp. application note 720007117, 2021.
(44) G. Isaac, I.D. Wilson, and R.S. Plumb, J. Chromatogr. A 1669, 462921 (2022).
(45) X. Liu, Q. Wang, and M.A. Lauber, J. Chromatogr. B 1191, 123120 (2022).
(46) T.H. Walter, B.A. Alden, K. Berthelette, J.A. Field, N.L. Lawrence, J. McLaughlin, and A.V. Patel, J. Sep. Sci. 45, 1389–1399 (2022).
(47) A. Lioupi, C. Virgiliou, T.H. Walter, K.M. Smith, P. Rainville, I.D. Wilson, G. Theodoridis, and H.G. Gika, J. Chromatogr. A 1672, 463013 (2022).
(48) D.V. McCalley, J. Chromatogr. A 1663, 462751 (2022).
(49) S. Fekete, M. DeLano, A. Bates-Harrison, S.J. Shiner, J.L. Belanger, K.D. Wyndham, and M.A. Lauber, Anal. Chem. 94, 3360–3367 (2022).
(50) J.M. Nguyen, M. DeLano, M.A. Lauber, and K. Wyndham, Waters Corp. application note 720007160, 2021.
(51) T. Arakawa, D. Ejima, T. Li, and J.S. Philo, J. Pharm. Sci. 99, 1674–1692 (2010).
(52) J.S. Philo, Curr. Pharm. Biotechnol. 10, 359–372 (2009).

 Thomas H. Walter, Bonnie A. Alden, Jonathan L. Belanger, Kenneth Berthelette, Cheryl Boissel, Mathew DeLano, Lavelay Kizekai, Jennifer M. Nguyen, and Stephen J. Shiner work in the Chemistry Technology Center at Waters Corporation, in Milford, Massachusetts. Direct correspondence to: tom_walter@waters.com.")
(From top left photo to bottom right) Thomas H. Walter, Bonnie A. Alden, Jonathan L. Belanger, Kenneth Berthelette, Cheryl Boissel, Mathew DeLano, Lavelay Kizekai, Jennifer M. Nguyen, and Stephen J. Shiner work in the Chemistry Technology Center at Waters Corporation, in Milford, Massachusetts. Direct correspondence to: tom_walter@waters.com.


















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


