Mixed-Mode Chromatography—A Review
Special Issues
Mixed-mode high performance liquid chromatography (MM-HPLC) involves the combined use of two (or more) retention mechanisms in a single chromatographic system. Many original stationary phases have been proposed in recent years with promising possibilities, while applications have only started to appear in the literature. In this review, the authors discuss mixed-mode chromatography stationary phases. An overview of applications using mixed-mode chromatography is described, as well as the increased interest in mixed-mode systems for two-dimensional chromatography.
Elise Lemasson1, Sophie Bertin2, Philippe Hennig2, Eric Lesellier1, and Caroline West1,1University of Orleans, CNRS, Institut de Chimie Organique et Analytique (ICOA), Orléans cedex, France, 2Institut de Recherches Servier, Suresnes, France
Mixed-mode high performance liquid chromatography (MM-HPLC) involves the combined use of two (or more) retention mechanisms in a single chromatographic system. Many original stationary phases have been proposed in recent years with promising possibilities, while applications have only started to appear in the literature. In this review, the authors discuss mixed-mode chromatography stationary phases. An overview of applications using mixed-mode chromatography is described, as well as the increased interest in mixed-mode systems for two-dimensional chromatography.
Among the numerous available separation modes, reversed-phase liquid chromatography (LC) is the favourite high performance liquid chromatography (HPLC) mode (1,2). In reversed-phase LC, C18 columns are most frequently utilized. With this kind of phase, one type of interaction dominates: the dispersion (London) interactions, also called hydrophobic interactions, between stationary phase and analytes. Secondary “parasite” hydrophilic interactions can appear between residual silanol groups and analytes, mainly when the phase is non-endcapped, which can lead to peak shape deformation and loss of efficiency. Reversed-phase LC is particularly suited for the separation of hydrophobic compounds, but fails for the retention of polar or charged compounds.
Different chromatographic modes must be envisaged as an alternative to reversed-phase LC for the analysis of such compounds. In the past, ionâpairing reversed-phase LC methods were developed to allow for the simultaneous retention of hydrophobic analytes and ions, for instance an active pharmaceutical ingredient (API) and its counterion. However, method development may be lengthy, and compatibility to mass spectrometry (MS) is not guaranteed (3). Hydrophilic interaction liquid chromatography (HILIC) (4) is an interesting alternative. In HILIC mode, the stationary phase used is hydrophilic and the mobile phase contains a high proportion of organic solvent (typically 70–98% acetonitrile, mixed with a buffer), which provides adequate retention for polar and ionic compounds and makes it compatible with electrospray ionization mass spectrometry (ESIâMS) (5–7). The retention mechanism is believed to be based on a combination of partitioning (between the mobile phase and a layer of water adsorbed on the stationary phase) and adsorption (onto the stationary phase surface or ligands). However, this technique also presents disadvantages: the low solubility of some compounds in high proportions of organic solvent, the long column equilibration time responsible for long method development time, and the critical influence of injection solvent on peak shape, retention, and separation make it impractical for open-access use (8,9). Ion-exchange chromatography (IEX) allows the separation of ionic compounds based on size and charge differences (10). However, the compatibility with MS can be difficult because of the high concentration of buffering salts in the mobile phase. Because of the high orthogonality between supercritical fluid chromatography (SFC) and reversedâphase LC, SFC can also be envisaged as an alternative (11–13). With the selectivity offered by the numerous types of stationary phases available in SFC, not only hydrophobic but also polar and ionizable solutes may be analyzed, with adjusted mobile phase conditions (14). Unfortunately, the majority of laboratories are equipped with HPLC systems. It is therefore easier to propose methods that would be compatible with HPLC systems to ensure their applicability.
In the 1980s, chromatographers started to investigate the use of secondary parasite interactions in reversed-phase LC to increase the separation power (15). The concept of mixed-mode liquid chromatography (MM-HPLC) as we know today appeared in 1986, when the combination of reversedâphase LC and IEX modes in one column was used for the separation of proteins (16). Since this first experiment, a variety of mixedâmode stationary phases have been developed, and some of them have been commercialized. In MM-HPLC, the stationary phases may be designed according to three different procedures: (i) one ligand is functionalized with different chemical functions (17,18); (ii) several different ligands are immobilized onto one support (17,19); or (iii) different groups or particles bonded with different types of ligands are mixed together in one column (20).
The intent of MM-HPLC is to use several retention mechanisms in a single column. MM-HPLC therefore shows great flexibility and versatility in the separation of various polar and nonâpolar compounds, owing to the multiple possibilities for interactions taking place between stationary phase and analytes (21). In addition, MM-HPLC is also highly compatible with MS because the concentrations of salts used are lower than in IEX mode.
In this review, selected particleâbased mixed-mode chromatography stationary phases are presented. An overview of applications with mixed-mode chromatography is also described. Finally, the interest of including MM-HPLC in 2D systems is discussed.
Mixed-Mode Chromatography Stationary Phases
This review is not intended to be exhaustive. Indeed, MM-HPLC stationary phases have been well detailed in previous reviews (22,23). This review article focuses solely on MM stationary phases developed for HPLC, and not for solidâphase extraction (SPE) purposes.
Judging from the types of interactions combined, mixed-mode stationary phases can be classified into four different groups: reversed-phase–hydrophilic interaction (reversed-phase LC–HILIC), reversedâphase–ion-exchange (reversed-phase LC–IEX), hydrophilic interaction–ion-exchange (HILIC–IEX), and tri-mode MM-HPLC (with several possible combinations). However, in practice, it often seems that only one retention mechanism can be used at the time, depending on the ratio of organic–aqueous portion in the mobile phase (20,24). Therefore, some of the stationary phases designed for MM-HPLC actually allow for multi-modal operation (for instance, reversedâphase LC or HILIC) but have not necessarily demonstrated combined mixedâmode mechanisms.
Reversed-Phase–Hydrophilic Interaction: Because reversed-phase LC is used for moderately polar and nonâpolar compounds and HILIC for polar compounds, the reversedâphase LC–HILIC mixed-mode presents the powerful advantage of retaining both hydrophobic and hydrophilic compounds. This combination should permit the analysis of complex mixtures with a wide range of polarities in a single run. The reversed-phase LC–HILIC stationary phases are designed by a combination of hydrophobic and polar groups. The hydrophobic portion is traditionally an alkyl chain or aromatic group. The chemical nature of the polar group varies between charge-neutral functions, like diol-, amide-, and cyano- or ionic-groups. In the latter case, the ionic functions are meant to support the aqueous pseudoâstationary phase of the HILIC retention mechanism, rather than an ion-exchange mechanism.
A typical reversed-phase LC–HILIC stationary phase is constituted of diol groups at the end of an alkyl chain (Figure 1[a]) (17,25). Aral et al. (26) designed a new stationary phase with two different amide groups, one was the terminal amide group and the other one was inserted between a phenyl ring and amino alcohol group (Figure 1[b]). This phase could sustain large variations of pH, which may be practical during method development, whenever ionizable species are concerned. Another type of stationary phase employed amino acids or short peptides as ligands. Li et al. developed a polyâL-lysine-grafted silica-based stationary phase (Figure 1[c]) (27). Lysine thus provides both a polar function (terminal amine group) and an alkyl chain. In a different paper, the authors directly immobilized a small peptide Boc-Phe-Aib-Phe-OH onto silica (28). This column demonstrated retention capability for various compounds, from hydrophobic compounds like polycyclic aromatic hydrocarbons or steroids to more polar compounds like nucleosides. In addition, the intrinsic chirality of the peptide ligand also imparted enantioselective capabilities to this phase. Surfactin, a peptide loop comprising seven amino acids and a β-hydroxyl fatty acid, was also employed as a mixed-mode ligand for reversed-phase LC–HILIC. Ohyama et al. (29) first reported the synthesis of a surfactinâmodifed silica stationary phase. With this phase, the retention of polar solutes depended on the acetonitrile content and exhibited a reversedâphase LC–HILIC mixedâmode retention behaviour.


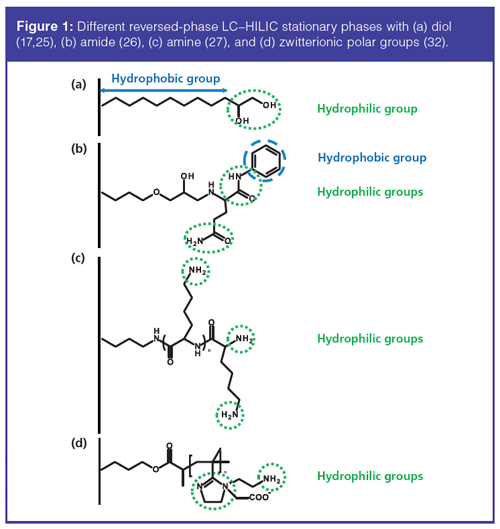
More recently, ionic liquids were used to develop zwitterionic ligands. The complex imidazolium organic cation and π conjugated system was commonly used to achieve the simultaneous separation of hydrophilic and hydrophobic compounds (30,31). Li et al. (32) used zwitterionic imidazoline to prepare a new type of stationary phase by polymerization on the silica surface with solvent-free microwaveâassisted organic synthesis (Figure 1[d]). With the use of ionic liquids, multiple interactions could take place: dipoleâdipole, electrostatic, π-π interaction, and hydrogen bonding. Qiao et al. (33) developed a new silica-based stationary phase with tricationic ionic liquid for the separation of flavonoids.
Reversed-Phase–Ion-Exchange: With reversed-phase LC–IEX mixed-mode, the retention of both hydrophobic and ionic compounds is achieved. The stationary phase ligands are constituted of an alkyl chain (C8 to C18) with ionic or ionizable groups at the end of, or embedded in the alkyl chain. In this kind of MM-HPLC, four IEX modes can be differentiated: strong cation-exchange (SCX), weak cation-exchange (WCX), strong anionâexchange (SAX), and weak anion-exchange (WAX).
For cation-exchange (CEX), similarly to traditional CEX stationary phases, acidic groups were generally used: carboxylic acid for WCX (Figure 2[a]) (34) and sulfonic acid for SCX (18) (Figure 2[b]). Zhang et al. (35) developed an original reversed-phase LC–WCX silicaâbased phase with a polystyrene network comprising carboxylic groups. For basic drugs separation, this phase exhibited good orthogonality with common C18 phases.

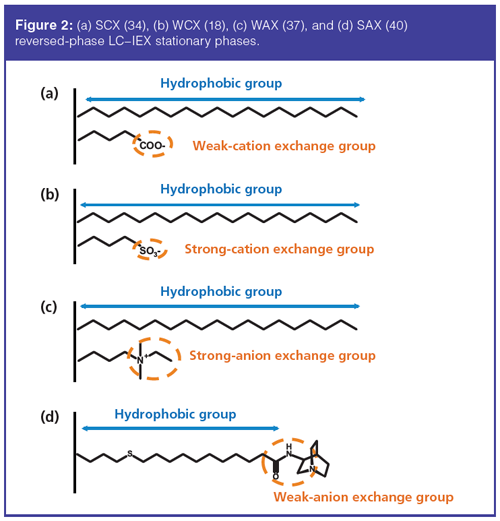
For anion exchange (AEX), quaternary ammonium can be used for SAX (36). Wei et al. (37) developed a new stationary phase with copolymerization of quaternary ammonium and C18 chain on silica (Figure 2[c]). For WAX, Lämmerhofer, Lindner, and co-workers developed a novel series of phases, a surfaceâbonded N-(10-undecenyl)-3-aminoquinuclidine silica-based stationary phase (38,39). This phase showed excellent performance for oligonucleotides separation (Figure 2[d]) (40). Recently, the same team developed novel stationary phases based on thiolâene click chemistry (41). These phases were synthesized by immobilization of N-undecenyl-3-α-aminotropane onto thiol-modified silica gel. The main advantages of theses phases with co-ionic endcapping were the strong reduction of retention times and the elution of acidic compounds with lower ionic strength, which enhanced the MS compatibility.
Once again, ionic liquids have been used to design imidazole-based stationary phases for reversed-phase LC–AEX (42). For example, Sun et al. (43,44) developed such phases for the separation of inorganic anions. The same team also synthesized more complex phases with dicationic imidazolium ionic liquids (45).
Hydrophilic Interaction–IonâExchange: The HILIC–IEX mode is adapted to the analysis of charged polar compounds. The main application field of this mode is the analysis of proteins or peptides. In the early 1990s, Zhu et al. analyzed peptides on strong cationâexchange columns (24). They first observed that IEX columns exhibited two different retention mechanisms depending on the acetonitrile percentage in the mobile phase. An IEX mechanism was observed at low percentage, while a HILIC-type retention mechanism was observed with increasing acetonitrile percentage.
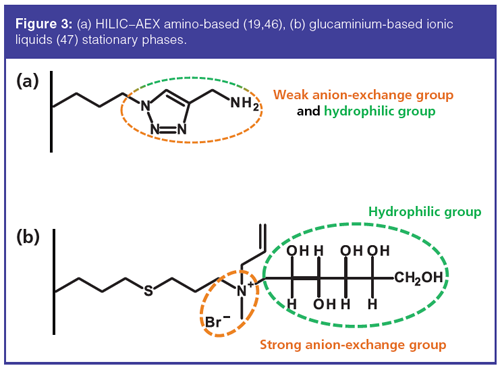
Later on, diverse stationary phases for HILIC–IEX mixed-mode were developed. The principle is always quite similar. Indeed, to obtain HILIC–IEX mixed-mode retention mechanisms, both charged (cationic, anionic, or zwitterionic) and uncharged hydrophilic groups are necessary. For HILIC–AEX mode, an amino group can be used as weak anion exchanger (Figure 3[a]) (19,46). Qiao et al. also used glucaminiumâbased ionic liquids (Figure 3[b]) for the separation of nucleosides in HILIC–SAX mode (47). Recently, Bo et al. (48) synthesized an original HILIC–IEC phase with adjustable selectivity by controlling the mixture ratio of two functional monomers. The application of the concept was demonstrated by the separation of nucleosides and β-agonists.

Tri-Mode MM-HPLC: As a result of the variety of analytes that may be encountered in a single sample, a need for increasingly varied interaction capabilities has arisen, and tri-mode MM-HPLC is being developed. While the simpler mixedâmode phases described above included the combination of two retention mechanisms, the triâmode stationary phases involve three different retention mechanisms. For this purpose, complex chemistries of stationary phases must be developed. The most common tri-mode used is the reversed-phase
LC–HILIC–AEX (49).
In 2012, Qiu et al. (21) developed a poly(ionic liquid)-grafted silica stationary phase, using ionic liquids. This phase allowed the effective retention of hydrophobic compounds, neutral polar molecules, and anions, owing to the presence of alkyl chain, carboxyl, and imidazolium groups, respectively (Figure 4[a]). In 2014, Li et al. (20) proposed a novel silicaâbased dendritic polymer reversed-phase LC–HILIC–AEX stationary phase composed of repeated patterns comprising phenyl ring, quaternary ammonium, and hydroxyl groups (Figure 4[b]). With this material, the use of the different retention mechanisms offered a wide range of selectivities. Indeed, under reversed-phase LC conditions, the authors separated polycyclic aromatic hydrocarbons (PAH). With the IEX mechanism, the analytes were separated in acids, bases, and neutrals compounds. Finally, under HILIC conditions, neutral amides were separated. The reversedâphase LC–HILIC–CEX mode was also described for small molecules analysis (50). The analysis of proteins can also be envisaged with this triâmode: Lin et al. (51) performed the separation of bovine serum albumin.

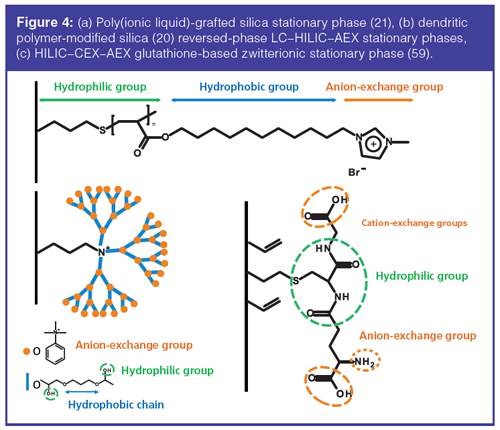
The range of applications of reversedâphase LC–AEX–CEX phases is large and allows the need for ionâpairing agents to be eliminated (52–55). Liu et al. (56) used a commercial tri-mode reversed-phase LC–AEX–CEX phase for the separation of pharmaceutical counterions. Another commercial triâmode HILIC–AEX–CEX was also used for the separation of pharmaceutical counterions (57). Qiao et al. (58) synthesized an original tri-mode phase with 4-Chloro-6-pyrimidinylferrocene modified silica gel for the separation of PAH, phenols, and aniline compounds. With this phase, reversed-phase LC, normal-phase, and AEX modes can all be envisaged. For HILIC–CEX–AEX, Shen et al. developed a glutathione-based zwitterionic stationary phase (59) for the separation of oligosaccharides. Glutathione is a tripeptide containing a free amino group and two carboxyl groups. With this peptide, there were two exchange sites available (Figure 4[c]). Wang et al. (60) used a multiâfunctionalized silica synthesized via “click chemistry”, which could be operated in tri-mode reversedâphase LC–CEX–HILIC, for the purification of quaternary ammonium alkaloids from plants.
One of the most complex mixedâmode phases in this category is the quinineâbased zwitterionic phase developed by the group of Lindner (61,62), now available as a commercial product. In addition to hydrophilic interaction, polar organic, anion-exchange, cationâexchange, and zwitterion-exchange retention mechanisms, enantioselectivity is also possible and was demonstrated with chiral separation of amino acids, small peptides, and acidic and basic analytes.
Finally, the tri-mode phases can be used for complex samples because of the large number of interactions involved. The challenge of tri-mode is to separate anionic, cationic, and neutral species by using only one column and one set of conditions. The choice of operating conditions remains primordial to take advantage of the wide selectivity offered
Use of Mixed-Mode HPLC for Achiral Applications
Pharmaceutical Applications: Pharmaceutical analyses (synthetic intermediates, APIs, impurities, or degradation products) are the most popular applications of mixed-mode chromatography.
Counterions analysis is very common in pharmaceuticals. Indeed, about 50% of pharmaceutical compounds are in the salt form, to improve their physicochemical properties like solubility, purity, and stability. Zhang et al. developed a method for the simultaneous separation of 25 counterions, including both cations and anions, organic and inorganic, in 20 min, using a gradient elution program with a reversed-phase LC–WAX–SCX tri-mode stationary phase (63). Liu et al. (56) used the same stationary phase to separate 10 counterions in isocratic elution. The same team also developed a method for the separation of other counterions using a HILIC–WCX–SAX stationary phase (57). In these papers, the authors also achieved the separation of basic and acidic APIs and their associated counterions. The European Pharmacopoeia (Ph. Eur.) validated the method developed by Zhang et al. for the separation of the 25 counterions in terms of specificity, repeatability, limits of quantification, and linearity. The Ph. Eur. demonstrated the applicability of the method for the identification and quantification of counterions in pharmaceuticals, coupled to an MS detector (64).
The identification and quantification of impurities in pharmaceutical products must be strictly controlled to ensure the efficiency and limited toxicity of the final product. MM-HPLC has proven its worth in the field of impurity profiling of small drugs (neutral polar or charged molecules) (65,66). Zhang et al. (35) used the reversed-phase LC–WCX phase described above for the separation of 43 basic drugs. This phase exhibited good orthogonality with common C18 phases. Strege et al. proposed the use of mixed-mode anion or cation-exchange–HILIC coupled to electron light scattering detection (ELSD)-ESI-MS as an alternative to reversed-phase LC for the analysis of small molecules in drug discovery (67). With the use of a mixed-mode column, the separation selectivity was highly orthogonal to reversed-phase LC. The authors also highlighted the advantage for preparative applications. Because of the high loadability of ion-exchange packings, the loading capacity was increased 10- to 100âfold in many cases in comparison to reversed-phase LC. This kind of mobile phase (acetonitrile gradient with 0.05% ammonium acetate and acetic acid) was also directly compatible with ELSD and MS detectors.
The families of compounds encountered in mixed-mode achiral analysis of drug-like compounds are varied: steroids (18,28), alkaloids (60,68), nonâsteroidal antiâinflammatory drugs (17,60,68,69), sulfonamides (70), nitroimidazoles (71), cannabinoids (72), water and fat-soluble vitamins (70,74–76), biphosphonates (77), and amphetamines (35). Analytical conditions (stationary and mobile phases, operating conditions) reported for the separation of families of drug compounds are detailed in Table 1.

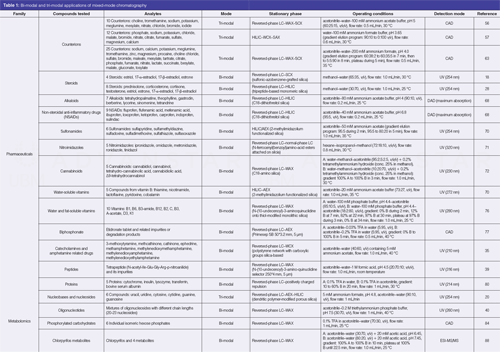
The use of peptides as therapeutics is fairly new (78). However, the range of therapeutic applications is steadily increasing: oncology, metabolic, cardiovascular diseases, and hematology are only a few examples. Lämmerhofer et al. (38) developed a reversedâphase LC–WAX stationary phase to separate molecules by lipophilicity and charge differences. The authors demonstrated the flexibility of retention and selectivity for peptide separations. Depending on the analytical conditions, different separation mechanisms were promoted: this stationary phase can be operated in reversedâphase LC, AEX, ionâexclusion, HILIC, and hydrophobic interaction chromatography (HIC) mode. Another mixed-mode reversed-phase LC–WAX stationary phase described above (with a N-(10-undecenoyl)â3-aminoquinuclidine selector [39]) was used as an alternative to a C18 stationary phase for the separation and purification of tetrapeptide and its side products. The authors demonstrated a better selectivity and enhanced loading capacity in comparison to reversed-phase LC.
MM-HPLC has also proven beneficial for the separation and purification of proteins. Published works have reviewed the development of mixed-mode ligands (86) constituted of aliphatic or aromatic groups as the hydrophobic part and an amino, carboxyl, or sulfonic group as the ionic moiety, and their applications in the biopolymer field (87). However, because proteins are complex molecules, the development of novel stationary phases with multiple interactions is needed to provide alternative selectivity to classical reversed-phase LC and hydrophobic interactions. The use of mixed-mode columns for proteomic purposes, such as the separation of therapeutic proteins, is also growing. Wang et al. developed a reversed-phase LC–IEX stationary phase based on ionic liquids. The authors demonstrated the high selectivity provided by this column, with the exclusion of basic proteins and the separation with good resolution of acidic ones (79). Ding et al. developed a reversedâphase LC–positively charged repulsion stationary phase, with the polymerization of polar group on the surface of silica (80). By adjusting operating conditions, the separation of five proteins was achieved. They also used the stationary phase to separate degradation products of one therapeutic protein, the recombinant human growth hormone.
Metabolomic Applications: Metabolomics studies require highly efficient, precise, and selective chromatographic methods. Among them, reversed-phase LC coupled to high resolution mass spectrometry (HRMS) is the most commonly used. However, metabolomics samples usually contain numerous analytes with a wide range of polarities. The use of mixed-mode columns is therefore advisable. Wernisch et al. (81) presented a comparison of the chromatographic performance of C18 reversed-phase LC, four HILIC, and two mixed-mode columns for the analysis of 764 metabolite standards, including amino acids, nucleotides, sugars, and other metabolites, representing all major biological pathways and commonly observed exogenous metabolites. The authors investigated retention capabilities, selectivities ,and specificity towards hydrophilic metabolites for each method. For phosphates, sugars, and amino acids, at least one of the mixedâmode columns permitted the retention of a higher percentage of metabolites with good peak shape than the C18 column. Ammann et al. (82) coupled two mixedâmode columns to obtain triâmodal reversedâphase LC, HILIC, and IEX separation capabilities in one run for the analysis of various metabolites: sugars, amino acids, carboxylates, fatty acids, and antioxidants. The authors also investigated the capability of a single tri-mode column for the separation of the same metabolites. The separation of 18 standards was partially achieved and the coupling with MS permitted the identification of metabolites.
Eastwood et al. (83) developed an analytical and semi-preparative method for the purification of nucleotides present in an enzymatic reaction mixture. The collected nucleotide was found to be pure at 99%. The dendritic polymer described above (20) was used for the effective separation of six nucleobases and nucleosides, with the combined use of reversed-phase LC, AEX, and HILIC interactions. Qiu et al. also developed a reversed-phase LC–AEX–HILIC stationary phase, based on ionic liquids for the separation of similar compounds. Zimmermann et al. (40) used a reversed-phase LC–WAX stationary phase based on N-(10-undecenoyl)-3-aminoquinuclidine for the separation of structurally closely related oligonucleotides, with a size of 20 to 23 nucleotides. Phosphorylated carbohydrates are essential metabolites for all forms of life. The difficulty in analysis is because there is a coexistence of isomers with a similar fragmentation in mass spectrometry and the complete resolution of isomers is difficult to obtain with reversedâphase LC. Hinterwirth et al. (84) developed a selective method for the separation of sugar phosphates by using a reversedâphase LC–WAX stationary phase. This phase revealed remarkable selectivities for the separation of six individual isomeric hexose phosphates, which were at least partially separated.
The use of mixed-mode columns has also appeared for lipidomic purposes. Granafei et al. proposed a method for the separation of a complex mixture containing polar lipids, with the use of a reversedâphase LC–HILIC mixedâmode liquid chromatography coupled to a high resolution tandem mass spectrometry system (85).
Mixed-mode material has also proven its application for the analysis of polar compounds in natural products. Apfelthaler et al. (86) evaluated the retention properties of 79 fungal metabolites on a reversedâphase LC–WAX mixed-mode stationary phase by LC–ESI-MS/MS. Qiao et al. (30) investigated the analysis of highly polar compounds with the analysis of secondary metabolites of Trichoderma, a genus of fungi, with an imidazoliumâembedded C8-based stationary phase for simultaneous reversedâphase LC–HILIC mixedâmode.
Finally, Bicker et al. (87) developed a method for the quantification of major chlorpyrifos metabolites, an insecticide, in human urine, by reversed-phase LC–WAX liquid chromatography coupled to ESI-MS/MS (88).
Some of these metabolomic applications are detailed in Table 1.
Interest of Mixed-Mode Columns in 2D Systems
The principle of two-dimensional liquid chromatography (2D-LC) is based on the combination of two different chromatographic systems, which may be provided by different stationary phases, different elution conditions, or different chromatographic modes.
Mixed-mode columns combine multiple chromatographic modes, which are complementary or orthogonal to each other. With such columns, the analysis of complex samples with only one column has been demonstrated (21,27) because of the great flexibility and high selectivity offered. The major advantage of using one mixedâmode column for two-dimensional separations is that it can replace the dual columns used in classical 2D-LC. Thus, the entire 2D-LC operation can be accomplished on a single column in off-line or on-line mode, by adjusting the analytical conditions to promote different retention mechanisms in the two dimensions.
Stevenson et al. (52) developed an off-line 2D-LC separation of a β-lactoglobulin tryptic digest with a single reversed-phase LC–AEX–CAX tri-mode stationary phase. In the first dimension, the mobile phase pH was 7, while it was adjusted to 2 in the second dimension. The authors also performed the same separation with a C18 column. Greater efficiency was observed for the mixed-mode column, thereby providing larger peak capacity than the C18 column.
Gilar et al. (89) developed a reversedâphase LC–CEX silicaâbased PFP column for the pseudo 2D-LC separation of peptides with strong negative moieties, such as phosphopeptides and sialylated glycopeptides. Recently, Wang et al. (17,90) synthesized a reversed-phase LC–HILIC C18-diol stationary phase for the analysis of complex samples of traditional Chinese medicines. They used their column for the simultaneous separation of highly polar and hydrophobic compounds in on-line and off-line 2D-LC, using a single column but changing the ratio of organic and buffer solution. Here again, the mixedâmode column showed enhanced peak capacity and higher efficiency when compared to a C18 column. Li et al. (91) analyzed urinary nucleosides in off-line 2D-LC with one reversed-phase LC–AEX phase (a silica stationary phase co-functionalized with Wulff-type phenylboronate and C12).
A mixed-mode column can also be coupled to a reversed-phase LC column for comprehensive 2D-LC analysis. Li et al. (92) used a reversedâphase LC–SAX in the first dimension and a reversedâphase C18 in the second dimension for the simultaneous separation of ionic and non-ionic compounds with different functional groups contained in white wine. With the use of a mixed-mode column in the first dimension, higher retention and larger peak distribution areas were obtained. Li et al. (93) used the same configuration for the analysis of polysorbate 20, a surfactant commonly used in the formulation of monoclonal antibodies to avoid protein denaturation and aggregation. In the first dimension, the mixedâmode column was used to separate polysorbate esters from the protein. Then, the esters were separated in the second dimension with the reversedâphase column. Finally, an offâline 2D mixed-mode reversed-phase LC–MS/MS method was developed for the profiling of lipids in biological samples. In the first dimension, lipids were separated according to their polarities on a monolithic silica-based mixed-mode column. In the second dimension, the separation was improved on a C30 core–shell particle phase. The method was applied to rat plasma and liver samples and more than 800 lipids were detected.
Conclusion and Perspectives
In this review, the recent development of mixed-mode chromatography stationary phases is covered. MM-HPLC provides unique flexibility because of the multiple retention mechanisms offered in one column. By adjusting the ratio of organic and aqueous phases and the concentration of aqueous buffers, reversed-phase LC, HILIC, and IEX modes can be successively used. The reversed-phase LC–HILIC, reversedâphase LC–IEX, HILIC–IEX, and tri-mode MM-HPLC are the most commonly encountered.
The field of applications of MM-HPLC is wide. MM-HPLC has demonstrated its efficiency for the combined analysis of small, non-polar, polar, and charged compounds, but also for larger molecules such as peptides or proteins. As a result, this technique is particularly used in pharmaceutical analysis for impurity profiling of APIs, counterions, drugs, vitamins, biopharmaceuticals, and much more. The high MS compatibility of mobile phases used with mixedâmode chromatography also permits the analysis of complex mixtures in metabolomics and lipidomics. For two-dimensional separations, one mixed-mode column can advantageously replace the dual columns used in classical 2D-LC. Thus, the entire 2D-LC operation may be accomplished on one single column in off-line or on-line modes, by adjusting the analytical conditions in the two dimensions. Compared to classical reversedâphase C18 phases, higher efficiency, peak capacity, and resolution were described. With the high flexibility offered by mixed-mode columns, a large number of molecules can be analyzed, making the possibilities of applications and the method development almost infinite.
References
- R. Nageswara Rao and V. Nagaraju, J. Pharm. Biomed. Anal.33, 335–377 (2003)doi:10.1016/S0731-7085(03)00293-0.
- S. Fekete, I. Molnár, and D. Guillarme, J. Pharm. Biomed. Anal.137, 60–69 (2017) doi:10.1016/j.jpba.2017.01.013.
- K. Petritis, S. Brussaux, S. Guenu, C. Elfakir, and M. Dreux, J. Chromatogr. A957, 173–185 (2002) doi:10.1016/S0021-9673(02)00372-2.
- A.J. Alpert, J. Chromatogr. A499, 177–196 (1990) doi:10.1016/S0021-9673(00)96972-3.
- D. Guillarme and J.-L. Veuthey, American Pharmaceutical Review20, (2017).
- B. Buszewski and S. Noga, Anal. Bioanal. Chem.402, 231–247 (2012) doi:10.1007/s00216-011-5308-5.
- A. Periat, J. Boccard, J.-L. Veuthey, S. Rudaz, and D. Guillarme, J. Chromatogr. A 1312, 49–57 (2013) doi:10.1016/j.chroma.2013.08.097.
- J. Ruta, S. Rudaz, D.V. McCalley, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A1217, 8230–8240 (2010) doi:10.1016/j.chroma.2010.10.106.
- B. Chauve, D. Guillarme, P. Cléon, and J.-L. Veuthey, J. Sep. Sci.33, 752–764 (2010) doi:10.1002/jssc.200900749.
- P. Cummins, K. Rochfort, and B. O’Connor, in Protein Chromatogr., D. Walls and S.T. Loughran, Eds. (Springer, New York, 2017) pp. 209–223. doi:10.1007/978-1-4939-6412-3_11.
- A. Grand-Guillaume Perrenoud, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A1266, 158–167 (2012) doi:10.1016/j.chroma.2012.10.005.
- E. Lemasson, S. Bertin, P. Hennig, H. Boiteux, E. Lesellier, and C. West, J. Chromatogr. A1408, 217–226 (2015) doi:10.1016/j.chroma.2015.07.037.
- E. Lemasson, S. Bertin, P. Hennig, H. Boiteux, E. Lesellier, and C. West, J. Chromatogr. A1408, 227–235 (2015) doi:10.1016/j.chroma.2015.07.035.
- E. Lesellier and C. West, J. Chromatogr. A 1382, 2–46 (2015) doi:10.1016/j.chroma.2014.12.083.
- A.P. Halfpenny and P.R. Brown, Chromatographia21, 317–320 (1986) doi:10.1007/BF02311602.
- L.A. Kennedy, W. Kopaciewicz, and F.E. Regnier, J. Chromatogr.359, 73–84 (1986).
- Q. Wang, M. Ye, L. Xu, and Z. Shi, Anal. Chim. Acta.888, 182–190 (2015) doi:10.1016/j.aca.2015.06.058.
- H. Qiu, M. Zhang, T. Gu, M. Takafuji, and H. Ihara, Chem.– Eur. J.19, 18004–18010 (2013) doi:10.1002/chem.201302746.
- Y. Liu, Q. Du, B. Yang, F. Zhang, C. Chu, X. Liang, Analyst 137, 1624–1628 (2012) doi:10.1039/C2AN16277F.
- Y. Li, J. Yang, J. Jin, X. Sun, L. Wang, and J. Chen, J. Chromatogr. A 1337, 133–139 (2014) doi:10.1016/j.chroma.2014.02.044.
- H. Qiu, A.K. Mallik, M. Takafuji, S. Jiang, and H. Ihara, Analyst 137, 2553–2555 (2012) doi:10.1039/C2AN35348B.
- L. Zhang, Q. Dai, X. Qiao, C. Yu, X. Qin, and H. Yan, TrAC Trends Anal. Chem.82, 143–163 (2016) doi:10.1016/j.trac.2016.05.011.
- L. Wang, W. Wei, Z. Xia, X. Jie, and Z.Z. Xia, TrAC Trends Anal. Chem.80, 495–506 (2016) doi:10.1016/j.trac.2016.04.001.
- B.-Y. Zhu, C.T. Mant, and R.S. Hodges, J. Chromatogr. A548, 13–24 (1991) doi:10.1016/S0021-9673(01)88590-3.
- X. Liu and C. Pohl, J. Chromatogr. A 1191, 83–89 (2008) doi:10.1016/j.chroma.2007.12.012.
- T. Aral, H. Aral, B. ZiyadanoÄulları, R. ZiyadanoÄulları, Talanta131, 64–73 (2015) doi:10.1016/j.talanta.2014.07.060.
- Y. Li, Z. Xu, Y. Feng, X. Liu, T. Chen, and H. Zhang, Chromatographia 74, 523 (2011) doi:10.1007/s10337-011-2120-5.
- S. Ray, M. Takafuji, and H. Ihara, J. Chromatogr. A1266, 43–52 (2012) doi:10.1016/j.chroma.2012.10.004.
- K. Ohyama, Y. Inoue, N. Kishikawa, and N. Kuroda, J. Chromatogr. A1371, 257–260 (2014) doi:10.1016/j.chroma.2014.10.073.
- X. Qiao, L. Zhang, N. Zhang, X. Wang, X. Qin, H. Yan, and H. Liu, J. Chromatogr. A 1400, 107–116 (2015) doi:10.1016/j.chroma.2015.04.060.
- S. Liu, H. Xu, J. Yu, D. Li, M. Li, X. Qiao, X. Qin, and H. Yan, Anal. Bioanal. Chem.407, 8989–8997 (2015) doi:10.1007/s00216-015-9064-9.
- Y. Li, Y. Feng, T. Chen, and H. Zhang, J. Chromatogr. A1218, 5987–5994 (2011) doi:10.1016/j.chroma.2011.04.023.
- L. Qiao, X. Shi, X. Lu, and G. Xu, J. Chromatogr. A1396, 62–71 (2015) doi:10.1016/j.chroma.2015.03.081.
- X. Cai, Z. Guo, X. Xue, J. Xu, X. Zhang, and X. Liang, J. Chromatogr. A1228, 242–249 (2012) doi:10.1016/j.chroma.2011.06.042.
- Y. Zhang and P.W. Carr, J. Chromatogr. A1218, 763–777 (2011) doi:10.1016/j.chroma.2010.11.009.
- A. Abbood, C. Smadja, C. Herrenknecht, Y. Alahmad, A. Tchapla, and M. Taverna, J. Chromatogr. A1216, 3244–3251 (2009) doi:10.1016/j.chroma.2009.02.031.
- J. Wei, Z. Guo, P. Zhang, F. Zhang, B. Yang, and X. Liang, J. Chromatogr. A 1246, 129–136 (2012) doi:10.1016/j.chroma.2012.03.047.
- M. Lämmerhofer, R. Nogueira, and W. Lindner, Anal. Bioanal. Chem. 400, 2517–2530 (2011) doi:10.1007/s00216-011-4755-3.
- R. Nogueira, M. Lämmerhofer, and W. Lindner, J. Chromatogr. A1089, 158–169 (2005) doi:10.1016/j.chroma.2005.06.093.
- A. Zimmermann, R. Greco, I. Walker, J. Horak, A. Cavazzini, and M. Lämmerhofer, J. Chromatogr. A1354, 43–55 (2014) doi:10.1016/j.chroma.2014.05.048.
- A. Zimmermann, J. Horak, O.L. Sánchez-Muñoz, and M. Lämmerhofer, J. Chromatogr. A 1409, 189–200 (2015) doi:10.1016/j.chroma.2015.07.036
- Y. Shan, L. Qiao, X. Shi, and G. Xu, J. Chromatogr. A1375, 101–109 (2015) doi:10.1016/j.chroma.2014.11.084.
- M. Sun, H. Qiu, L. Wang, X. Liu, and S. Jiang, J. Chromatogr. A1216, 3904–3909 (2009) doi:10.1016/j.chroma.2009.02.089.
- M. Sun, J. Feng, C. Luo, X. Liu, and S. Jiang, Talanta105, 135–141 (2013) doi:10.1016/j.talanta.2012.11.077.
- M. Sun, J. Feng, X. Wang, H. Duan, L. Li, and C. Luo, J. Sep. Sci.37, 2153–2159 (2014) doi:10.1002/jssc.201400176.
- G. Shen, F. Zhang, B. Yang, C. Chu, and X. Liang, Talanta115, 129–132 (2013) doi:10.1016/j.talanta.2013.04.046.
- L. Qiao, S. Wang, H. Li, Y. Shan, A. Dou, X. Shi, and G. Xu, J. Chromatogr. A1360, 240–247 (2014) doi:10.1016/j.chroma.2014.07.096.
- C. Bo, X. Wang, C. Wang, and Y. Wei, J. Chromatogr. A1487, 201–210 (2017) doi:10.1016/j.chroma.2017.01.061.
- P. Zhang, J. Chen, and L. Jia, J. Chromatogr. A1218, 3459–3465 (2011) doi:10.1016/j.chroma.2011.03.062.
- H. Huang, Z. Lin, Y. Lin, X. Sun, Y. Xie, L. Zhang, and G. Chen, J. Chromatogr. A1251, 82–90 (2012) doi:10.1016/j.chroma.2012.06.032.
- Z. Lin, X. Tan, R. Yu, J. Lin, X. Yin, L. Zhang, and H. Yang, J. Chromatogr. A1355, 228–237 (2014) doi:10.1016/j.chroma.2014.06.023.
- P.G. Stevenson, J.N. Fairchild, and G. Guiochon, J. Chromatogr. A1218, (2011) 1822–1827. doi:10.1016/j.chroma.2011.01.078.
- M. Biba, E. Jiang, B. Mao, D. Zewge, J.P. Foley, and C.J. Welch, J. Chromatogr. A1304, 69–77 (2013) doi:10.1016/j.chroma.2013.06.050.
- A.A. Kazarian, P.N. Nesterenko, P. Soisungnoen, R. Burakham, S. Srijaranai, and B. Paull, J. Sep. Sci. 37, 2138–2144 (2014) doi:10.1002/jssc.201400411.
- H. Choi, S. Kim, S. Ahn, H. Chang, S. Lee, and Y. Lee, Forensic Sci. Int.259, 69–76 (2016) doi:10.1016/j.forsciint.2015.12.016.
- X. Liu and C.A. Pohl, J. Chromatogr. A1232, 190–195 (2012) doi:10.1016/j.chroma.2011.12.009.
- X. Liu, M. Tracy, U. Aich, and C. Pohl, “Exploring Mixed-Mode Chromatography: Column Chemistry, Properties, and Applications”, (2014). https://tools.thermofisher.com/content/sfs/posters/PN-20947-Mixed-Mode-Chromatography-HPLC-2014-PN20947-EN.pdf (accessed 5 April 2017).
- L. Qiao, X. Zhou, Y. Zhang, A. Yu, K. Hu, S. Zhang, and Y. Wu, Talanta153, 8–16 (2016) doi:10.1016/j.talanta.2016.02.055.
- A. Shen, X. Li, X. Dong, J. Wei, Z. Guo, and X. Liang, J. Chromatogr. A1314, 63–69 (2013) doi:10.1016/j.chroma.2013.09.002.
- Q. Wang, Y. Long, L. Yao, M. Ye, and L. Xu, Phytochem. Anal. (2017) doi:10.1002/pca.2680.
- F. Ianni, R. Sardella, A. Carotti, B. Natalini, W. Lindner, and M. Lämmerhofer, Chirality28, 5–16 (2016) doi:10.1002/chir.22541.
- C.V. Hoffmann, R. Reischl, N.M. Maier, M. Lämmerhofer, and W. Lindner, J. Chromatogr. A 1216, 1147–1156 (2009) doi:10.1016/j.chroma.2008.12.045.
- K. Zhang, L. Dai, N.P. Chetwyn, J. Chromatogr. A1217, 5776–5784 (2010) doi:10.1016/j.chroma.2010.07.035.
- D. Ilko, C.J. Nap, U. Holzgrabe, and S. Almeling, Pharmeuropa Bio Sci. Notes 2014, 81–91 (2014).
- J. Li, S. Shao, M.S. Jaworsky, and P.T. Kurtulik, J. Chromatogr. A1185, 185–193 (2008) doi:10.1016/j.chroma.2008.01.083.
- B.A. Olsen, J. Chromatogr. A913, 113–122 (2001) doi:10.1016/S0021-9673(00)01063-3.
- M.A. Strege, S. Stevenson, and S.M. Lawrence, Anal. Chem.72, 4629–4633 (2000).
- Q. Wang, Y. Long, L. Yao, L. Xu, Z.-G. Shi, and L. Xu, Talanta146, 442–451 (2016) doi:10.1016/j.talanta.2015.09.009.
- Q. Wang, L. Xu, and Y. Xue, J. Liq. Chromatogr. Relat. Technol. 39, 598–606 (2016) doi:10.1080/10826076.2016.1213174.
- B. Yang, H. Liu, J. Chen, M. Guan, and H. Qiu, J. Chromatogr. A 1468, 79–85 (2016) doi:10.1016/j.chroma.2016.09.021.
- X. Zhou, X. Li, A. Cao, Q. Lijun, A. Yu, S. Zhang, and Y. Wu, Talanta144, 1044–1051 (2015) doi:10.1016/j.talanta.2015.07.063.
- C.-H. Hung, J. Zukowski, D.S. Jensen, A.J. Miles, C. Sulak, A.E. Dadson, and M.R. Linford, J. Sep. Sci.38, 2968–2974 (2015) doi:10.1002/jssc.201500156.
- D.W. Later, B.E. Richter, D.E. Knowles, and M.R. Andersen, J. Chromatogr. Sci.24, 249–253 (1986) doi:10.1093/chromsci/24.6.249.
- M. Lämmerhofer, M. Richter, J. Wu, R. Nogueira, W. Bicker, and W. Lindner, J. Sep. Sci. 31, 2572–2588 (2008) doi:10.1002/jssc.200800178
- H. Wang, L. Zhang, T. Ma, L. Zhang, and X. Qiao, J. Sep. Sci. 39, 3498–3504 (2016) doi:10.1002/jssc.201600448.
- R. Dabre, N. Azad, A. Schwämmle, M. Lämmerhofer, and W. Lindner, J. Sep. Sci.34, 761–772 (2011) doi:10.1002/jssc.201000793.
- X.-K. Liu, J.B. Fang, N. Cauchon, and P. Zhou, J. Pharm. Biomed. Anal.46, 639–644 (2008) doi:10.1016/j.jpba.2007.11.041.
- K. Fosgerau and T. Hoffmann, Drug Discov. Today 20, 122–128 (2015) doi:10.1016/j.drudis.2014.10.003.
- Y.-X. Wang, K.-L. Zhao, F. Yang, L. Tian, Y. Yang, and Q. Bai, Chin. Chem. Lett.26, 988–992 (2015) doi:10.1016/j.cclet.2015.05.001.
- L. Ding, Z. Guo, Z. Hu, and X. Liang, J. Pharm. Biomed. Anal.138, 63–69 (2017) doi:10.1016/j.jpba.2017.01.004.
- S. Wernisch, R. Pell, and W. Lindner, J. Sep. Sci. 35, 1560–1572 (2012) doi:10.1002/jssc.201200103.
- A.A. Ammann and M.J.-F. Suter, J. Chromatogr. A. 1456, 145–151 (2016) doi:10.1016/j.chroma.2016.06.001.
- H. Eastwood, F. Xia, M.-C. Lo, J. Zhou, J.B. Jordan, J. McCarter, W.W. Barnhart, and K.-H. Gahm, J. Pharm. Biomed. Anal.115, 402–409 (2015) doi:10.1016/j.jpba.2015.08.001.
- H. Hinterwirth, M. Lämmerhofer, B. Preinerstorfer, A. Gargano, R. Reischl, W. Bicker, O. Trapp, L. Brecker, and W. Lindner, J. Sep. Sci. 33, 3273–3282 (2010) doi:10.1002/jssc.201000412.
- S. Granafei, P. Azzone, V.A. Spinelli, I. Losito, F. Palmisano, and T.R.I. Cataldi, J. Chromatogr. A 1477, 47–55 (2016) doi:10.1016/j.chroma.2016.11.048.
- E. Apfelthaler, W. Bicker, M. Lämmerhofer, M. Sulyok, R. Krska, W. Lindner, and R. Schuhmacher, J. Chromatogr. A1191, 171–181 (2008) doi:10.1016/j.chroma.2007.12.067.
- W. Bicker, M. Lämmerhofer, and W. Lindner, Anal. Bioanal. Chem.390, 263–266 (2008) doi:10.1007/s00216-007-1637-9.
- W. Bicker, M. Lämmerhofer, and W. Lindner, J. Chromatogr. B822, 160–169 (2005) doi:10.1016/j.jchromb.2005.06.003.
- M. Gilar, Y.-Q. Yu, J. Ahn, J. Fournier, and J.C. Gebler, J. Chromatogr. A 1191, 162–170 (2008) doi:10.1016/j.chroma.2008.01.061.
- Q. Wang, L. Tong, L. Yao, P. Zhang, and L. Xu, J. Pharm. Biomed. Anal.125, 205–211 (2016) doi:10.1016/j.jpba.2016.03.033.
- H. Li, X. Zhang, L. Zhang, X. Wang, F. Kong, D. Fan, L. Li, and W. Wang, Anal. Chim. Acta.962, 104–113 (2017) doi:10.1016/j.aca.2017.01.028.
- D. Li, R. Dück, and O.J. Schmitz, J. Chromatogr. A1358, 128–135 (2014) doi:10.1016/j.chroma.2014.06.086.
- Y. Li, D. Hewitt, Y.K. Lentz, J.A. Ji, T.Y. Zhang, and K. Zhang, Anal. Chem.86, 5150–5157 (2014) doi:10.1021/ac5009628.
Elise Lemasson is a PhD student at the University of Orleans, France. Her research focuses on developing generic methods in HPLC and SFC for impurity profiling of drug candidates.
Sophie Bertin is a senior scientist at Servier Research Institute, specializing in separation science and mass spectrometry.
Philippe Hennig is the head of the analytical department of Servier Research Institute.
Eric Lesellier is an Associate Professor in analytical chemistry, teaching at Paris-Orsay University and carrying out his research at the University of Orleans. He is a recognized expert in chromatographic stationary phases for HPLC and SFC. He has contributed significantly to developing fundamental knowledge and applications of SFC.
Caroline West is an Associate Professor in analytical chemistry at the University of Orleans. Her scientific interests lie in fundamentals of chromatographic selectivity, both in the achiral and chiral modes, mainly in SFC but also in HPLC. In 2015, she received the LCGC award for “Emerging Leader in Chromatography”.







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Altering Capillary Gas Chromatography Systems Using Silicon Pneumatic Microvalves
May 5th 2025Many multi-column gas chromatography systems use two-position multi-port switching valves, which can suffer from delays in valve switching. Shimadzu researchers aimed to create a new sampling and switching module for these systems.


Studying Cyclodextrins with UHPLC-MS/MS
May 5th 2025Saba Aslani from the University of Texas at Arlington spoke to LCGC International about a collaborative project with Northwestern University, the University of Hong Kong, and BioTools, Inc., investigating mirror-image cyclodextrins using ultra-high performance liquid chromatography–tandem mass spectrometry (UHPLC–MS/MS) and vibrational circular dichroism (VCD).



