Computer-Assisted Method Development for Small and Large Molecules
Special Issues
The aim of this article is to illustrate the current status of computer-assisted method development and retention modelling. This study focuses on the successful method development of typical small pharmaceutical compounds (impurity profiling) and large therapeutic proteins. By choosing appropriate initial conditions, the method development can be performed in less than one day. However, for small molecules possessing different physicochemical properties, the conditions can be multifarious, while for biopharmaceuticals (for example, monoclonal antibodies [mAbs], antibody–drug conjugates [ADCs]), a generic method can easily be developed. In addition to retention modelling and optimization, the potential of simulated robustness testing is also demonstrated. Depending on the applied retention model, the impact of any change among six experimental parameters (tG, T, pH, ternary composition, flow rate, and initial- and final mobile phase compositions) on the separation can be assessed using a 26 or 36 type virtual
Szabolcs Fekete1, Róbert Kormány2, and Davy Guillarme1, 1School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Geneva, Switzerland, 2EGIS Pharmaceuticals Plc., Budapest, Hungary
The aim of this article is to illustrate the current status of computer-assisted method development and retention modelling. This study focuses on the successful method development of typical small pharmaceutical compounds (impurity profiling) and large therapeutic proteins. By choosing appropriate initial conditions, the method development can be performed in less than one day. However, for small molecules possessing different physicochemical properties, the conditions can be multifarious, while for biopharmaceuticals (for example, monoclonal antibodies [mAbs], antibody–drug conjugates [ADCs]), a generic method can easily be developed. In addition to retention modelling and optimization, the potential of simulated robustness testing is also demonstrated. Depending on the applied retention model, the impact of any change among six experimental parameters (tG, T, pH, ternary composition, flow rate, and initial- and final mobile phase compositions) on the separation can be assessed using a 26 or 36 type virtual factorial design. No additional experiments are required to perform the robustness evaluation. Finally, virtual method transfer between different chromatographic systems is demonstrated.
Method development in liquid chromatography (LC) encompasses the search for the optimal operating conditions (type of mobile and stationary phase, temperature, gradient steepness, pH, ionic strength) resulting in the separation of a mixture into its individual constituents (1). Because of the high probability of peak overlap in complex mixtures and the dependence of retention and selectivity on the chromatographic parameters used, the method development process is often tedious and time-consuming (up to several weeks of work) (2). Knowledge and expertise are required from the chromatographer and a lot of trial-and-error type processes are still involved. Software-assisted retention modelling and method optimization offer the potential to speed up the overall process. The gain in method development time can be particularly interesting for large molecules (peptides and proteins), which are much more affected by the change in solvent strength than small molecules and hence often require very shallow and long gradients (on–off retention mechanism) (1). The development of such long gradients without retention modelling is therefore iterative and time-consuming (3).
In an effort to improve the efficiency of method development and increase information about method specificity, several computer modelling programs have been developed in the last 25 years. One strategy is based on the optimization of the design space by measuring and visualizing the effect of the mobile phase composition (gradient time and shape, pH, ionic strength, ternary eluent, additive concentrations, and temperature) on a given stationary phase. Another process is based on the use of a customized database or vendor database of chromatographic methods, where method conditions can be predicted from compound structures. Expert systems predict physicochemical properties (pKa, log P, logD) of the solutes and suggest a mobile phase composition for the separation.
The combination of the three approaches is also feasible and enables an automated strategy for high performance liquid chromatography (HPLC) method development as reported by Galushko et al. (4). Krisko et al. presented a strategy that uses an automated column selection system and a series of HPLC columns with different hydrophobicity and silanol activity, in combination with modelling software to successfully develop chromatographic methods (5).
The aim of this article is to illustrate the current possibilities of softwareâassisted HPLC method development by showing representative pharmaceutical applications.
Impurity and Degradation Profiling of Small MoleculeâBased Pharmaceuticals
It has already been recommended since the 1990s to perform a few numbers of initial gradient runs (for example, 4 runs) for multifactorial (two-dimensional [2D]) experimental designs (6). A typical approach is to simultaneously model the evolution of selectivity (resolution) as a function of mobile phase temperature and gradient steepness on a selected column. In this case, the required ”scouting” runs are those experiments on which the computer models are “based” (calculated) to model a few thousand experiments with an accuracy of around 99%. Now, thanks to the development of modelling software, three-dimensional (3D) resolution cubes can also be easily built based on 6–12 experiments (7). The most important method variables for such a 3D model are typically the gradient steepness, mobile phase temperature, and mobile phase pH or sometimes ternary composition, but there is also the possibility to select other factors such as additive concentration and ionic strength.
For the impurity profiling of small molecule pharmaceuticals, the pharmacopeia methods still have to be considered as references. Unfortunately, pharmacopeia monographs often describe and suggest obsolete, time-consuming conventional HPLC separations-or even thin layer chromatographic (TLC) methods. Here, this article illustrates the renewal of such a pharmacopeia method using state-of-the-art retention modelling and ultrahigh-pressure liquid chromatography (UHPLC) technology.
An old-fashioned method for terazosin has been recently reviewed and optimized to reduce the analysis time from 90 min (2 × 45 min) to 5 min (8). The European Pharmacopoeia (Ph. Eur.) method determines the specified impurities separately using two different methods. The reason for using two methods is that the retention of two impurities is not sufficient in common reversed phase conditions, even with 100% water as the mobile phase. Therefore ion-pair chromatography is suggested. In addition, two impurities absorb at low ultraviolet (UV)âwavelength and have therefore to be acquired at a different wavelength. This way, the Ph. Eur. method becomes unnecessarily complicated and timeâconsuming.
By working at high mobile phase pH, the retention of the poorly retained terazosin impurities can be sufficiently enhanced. High pH separations can now be performed on hybrid stationary phases, which were probably not available when the Ph. Eur. method was developed. In addition to high pH separation, an appropriate detection wavelength can also be selected for all terazosin impurities.
After a pH screening study, it was shown that pH > 10 provided sufficient retention for all terazosin impurities. The use of 0.1% v/v NH4OH-solution was found to be optimal to avoid additive precipitation in organic modifiers. Since the type of organic modifier had a significant effect on selectivity and resolution between impurities, a gradient-time–temperature–ternary composition (tG × T × tc) model was built on the basis of 12 initial runs. The impact of gradient steepness was studied at two levels (tG1 = 3 min, tG2 = 9 min), temperature was set to T1 = 20 ºC and T2 = 40 ºC, while the organic modifier in mobile phase B was changed as tc1: 100% acetonitrile, tc2: 50% acetonitrile + 50% methanol, and tc3: 100% methanol. The 50 × 2.1 mm C18 column was operated at 0.5 mL/min. Figure 1 shows the developed resolution cube (design space) and critical resolution planes corresponding to (a) 100% acetonitrile, (b) 50% acetonitrile in methanol, and (c) 100% methanol as strong mobile phase.


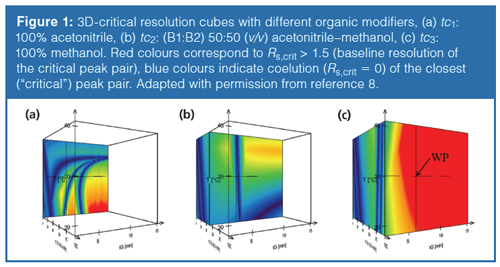
As shown in Figure 1, the protic solvent (methanol) provided higher resolution and more robust separation than acetonitrile. Figure 2 shows (a) the predicted and (b) the experimentally verified chromatograms when performing the separation at the selected working point (WP, see Figure 1[c]). The predicted and observed retention times, as well as resolution values, were in excellent agreement. The average deviation of the predicted retention times was less than 1 s compared to the measured ones, and the critical resolution (Rs,crit) was 1.82 in both cases.

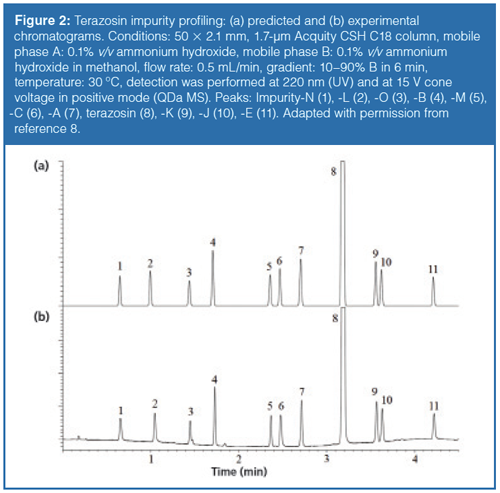
Finally, the original 90-min long analysis could be cut down to only 5 min by using a 50 × 2.1 mm UHPLC column instead of a conventional HPLC column. The total method development time was no longer than three days as a result of the small column dimensions.
Simulated Robustness Testing
A fundamental criteria of quality in HPLC separations is robustness. Guidelines define the robustness of an analytical procedure as “a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters...” providing “...an indication of its reliability during normal usage” (9). Historically, robustness testing was performed as the final step of a method development process, during the validation stage, which often led to unexpected observations. However, because a method considered as non-robust should be redeveloped and revalidated, robustness should be verified earlier in the method lifetime, that is, at the method development stage or at the beginning of the validation procedure (10,11). Generally, two approaches are used for robustness testing, according to the ICH definition in pharmaceutical analytical practice. Either a oneâfactorâat-a-time (OFAT) procedure or an experimental design (DoE) procedure could be applied. Both approaches require additional experiments and can sometimes be time-consuming.
A useful feature of current modelling software is the possibility to perform an in-depth “modelled” robustness testing, without the need for additional experiments. From the design space, as defined in a resolution map or cube, it is possible to get robustness information for the measured parameters (tG, T, tc, or pH). In addition, the influence of additional factors, such as flowrate or initial and final mobile phase composition of a gradient, on resolution can also be estimated. Consequently, the impact of changes in any of the six parameters on the resolution can be assessed using a simulated 26 or 36 type factorial design (12). The possible deviations from the nominal values have to be defined (input in the software) and then the software simulates 64 or 729 conditions (depending on the type of factorial design). At the end, a “frequency distribution graph” can be obtained that shows the probability of fulfilling a given resolution criteria under any combination of possible parameters. On the other hand, “regression coefficients” can also be obtained to show the effect of each parameter (and their combinations), related to the selected deviation from the nominal value, for the critical resolution. The reliability of such a virtual robustness test has been studied and compared to real experiments based on DoE robustness tests (12). It appeared that the retention time relative errors were maximally 4% (lower than 1% for the average relative error). On the other hand, the predicted critical resolution errors were between 6.9 and 17.2%. This accuracy of virtual robustness test is more than acceptable and enables robustness studies to be developed at the very beginning of the method development.
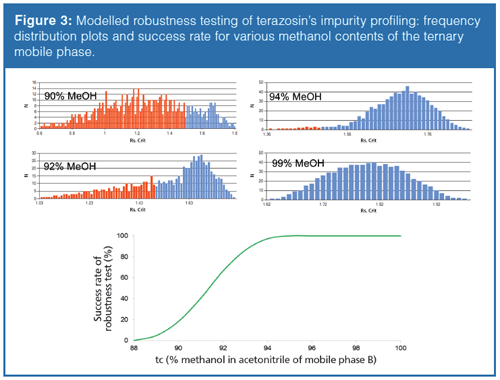
To illustrate the usefulness of simulated robustness tests, the previous example of terazosin impurity profiling was considered (section 2). Since the ternary mobile phase composition seemed to be a critical factor, it is worth performing robustness studies at different ternary compositions to prove that the most robust working point (100% methanol as organic modifier) was selected. For this illustration, the following mobile phase compositions were considered close to the working point: tc1: 90%, tc2: 92%, tc3: 94%, and tc4: 99% methanol in mobile phase B. The required resolution was Rs = 1.5. Besides the three model variables (tG, T, and tc), the flow rate-as well as initial and final composition of the mobile phase-was also included in the built up 36 design. Then, the 729 experiments were simulated. Figure 3 shows the obtained frequency distributions for the different methanol fractions and the success rate of robustness test as a function of methanol. On the frequency distribution plots, the red bars correspond to the experiments that did not fulfil the Rs≥1.5 criteria. It can clearly be seen that the higher the methanol content in mobile phase B, the higher the success rate of the robustness testing is. This suggests that 100% methanol as organic modifier was a good choice because it provides a robust working point.

Method Transfer
To increase sample throughput, a conventional HPLC analysis can be transferred to UHPLC. Alternatively, to decrease method development time, a separation can be developed in UHPLC conditions and then transferred to conventional HPLC for routine analysis. In both cases, when transferring an analysis from conventional HPLC to UHPLC-or vice-versa-comparable method parameters must be used to maintain equivalent separations.
To have a method compatible with any column dimensions and HPLC or UHPLC instruments, the optimized methods can virtually be transferred to other columns of different lengths, inner diameters, and particle sizes, and for various system gradient delays and extra-column volumes. By utilizing HPLC modelling software, it is possible to automatically calculate and predict the impact of column parameters (length, diameter, particle size), system dwell volume, and extra-column volume on the resolution. Moreover, experimentally observed column porosity can also be considered for making the transfer more accurate (porosity of HPLC and UHPLC columns is often slightly different). For such a simulated method transfer, the initial data acquired on a given column and system have to be changed virtually and the geometrical method transfer rules have to be considered (13). The accuracy of UHPLC to HPLC method transfer was found to be excellent in previous studies (13).
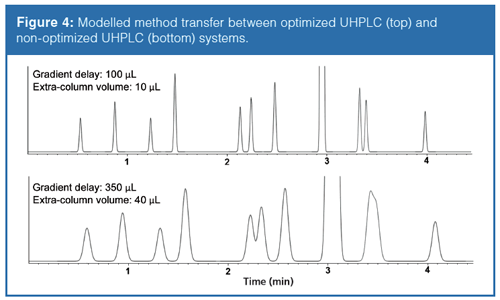
This article illustrates the transfer between different LC systems when using very efficient 50 × 2.1 mm columns. Often when a UHPLC method is developed in a research laboratory and then transferred to the QC laboratory the UHPLC system is different (different provider, different extra-column volume, binary or quaternary pumps). Again, the terazosin separation was taken as an example. The method mentioned earlier was developed on an optimized UHPLC system, possessing a 100âµL gradient delay (dwell) volume (binary pumping system) and around 10-µL extra-column volume (0.065 mm i.d. connector tubes). The goal of this study was to highlight what happens when using this method on a nonâoptimized UHPLC system possessing a 350-µL gradient delay volume (quaternary pumping system) and a 40-µL extra-column volume (0.125-mm i.d. connector tubes). Figure 4 shows the simulated separations on the two different UHPLC systems. As expected, there is a systematic shift in the retention times in proportion with the difference in gradient delay volumes. In addition-because of the differences in extra-column volumes-the apparent efficiency of the separation performed on the non-optimized system decreased drastically. By performing such a virtual system transfer, surprises during method transfer between different laboratories can be avoided.

Analysis of Therapeutic Proteins
In contrast to small molecules, large molecules such as proteins show different retention mechanisms in several modes of chromatography such as: (i) an on–off mechanism retaining the macromolecules at the column inlet until at some point in the gradient they are desorbed and then move through the column without any further interaction; (ii) precipitationâredissolution, that is, separation based on solubility instead of interaction with the stationary phase; and (iii) multipoint attachment to the surface of the stationary phase (1).
While these mechanisms are fundamentally different from those observed with small molecules, the gradient separation of macromolecules can still be predicted from the linear solvent strength (LSS) theory or from a slightly modified model. The reason is that, in most cases, a relatively limited range of the method variables has to be studied because sufficient retention, recovery, and peak shape can only be obtained in a limited design space. As an example, for monoclonal antibodies (mAbs) in reversed-phase mode, the temperature has to be kept between 70 °C and 90 °C to get acceptable recovery and peak shape. Therefore, performing measurements at lower temperatures makes no sense. It is known that mAbs show deviations from the common linear Van’t Hoff type behaviour (temperature dependency of the retention) in a wide temperature range, as a result of possible conformational changes. It was also shown that in a narrow temperature range (for example, ΔT = 20 ºC), a linear retention model provides excellent prediction accuracy (14). The situation is similar with organic modifier, ion-pairing reagent, and pH because only a limited range has to be studied because of the on–off retention mechanism of proteins. In such a narrow range of method variables, simple linear models (or polynomial ones) can be used.
With large molecules-in most cases-generic conditions can be applied for different protein classes (for example, cytokines, mAbs, antibody–drug conjugates [ADCs]). Indeed, the structure of the different proteins within a class is very similar. The amino acid sequence is very close and the global conformation is similar. It is also clear that the variants, which have to be separated from the native protein and from each other, possess relatively small differences compared to the native protein (such as the oxidation of some amino acids, deamidation, reduction of a disulfide bond). In a whole protein structure, those modifications are minor compared to the native amino acid sequence (for example, modifications of 2–5 amino acids from the total few hundred or thousand amino acids in the protein backbone).
To conclude on proteins HPLC method development, generic conditions can be used for the optimization in most cases and the impact of method variables on the separation has to be studied only in a narrow range.
Reversed-Phase Liquid Chromatography: Reversed phase chromatography is a denaturating mode and offers very efficient separations for large proteins. It is historically used to separate hydrophobic variants of a given protein in harsh conditions (low pH, organic modifier, elevated temperature).
The most useful retention models can be based on (i) gradient steepness–temperature (tG × T), or on (ii) gradient steepness–temperature–ternary mobile phase composition (tG × T × tc) models.
In the following example, a fractional design, consisting of 2 × 2 × 3 experiments, was applied for a reduced cysteine-linked IgG1 ADC. The conventional, smallâmolecule linear relationship was used to model the gradient steepness and temperature, while the ternary composition was modelled using a quadratic relationship. For each of the three experimental sets (ternary composition), two-dimensional resolution maps (gradient time against temperature) were generated. They were used to create a threeâdimensional resolution cube to visualize the combined influence of the optimization parameters. Figure 5 represents a resolution cube, based on three method variables, and the chromatogram at the working point. The part of the ”design space” where Rs > 1 is shown in red.

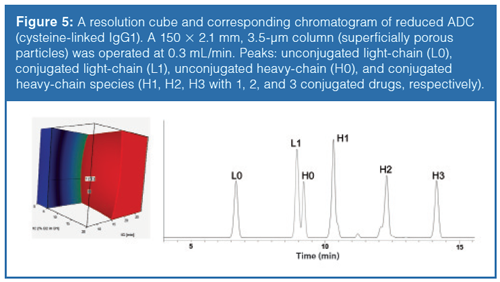
The accuracy of retention time and resolution predictions in reversedâphase LC mode were excellent for large proteins (digested mAbs, reduced mAbs, ADCs, reduced ADCs), as reported elsewhere (1,14,15).
Ion Exchange Chromatography (IEX): IEX is an historical and nonâdenaturing chromatographic mode considered as a reference technique for the qualitative and quantitative evaluation of charge heterogeneity of therapeutic proteins. Among the different IEX modes, cation-exchange chromatography (CEX) is the most widely used for protein characterization because most biopharmaceuticals (mAbs) possess a high isoelectric point (pI). CEX is considered as the gold standard for charge variants analysis, but method parameters, such as column type, mobile phase pH, and salt concentration gradient, often need to be optimized.
In classical IEX, a linear saltâgradient is applied for the elution and a constant mobile phase pH is maintained. Recently, however, pH gradient mode has regained interest, since generic, multiproduct methods can be developed.
The most efficient design space in IEX can be based on (i) gradient steepness–pH (tG × pH) model in salt gradient mode or on (ii) gradient steepness–temperature (tG × T) model in pH gradient mode.
Computer-assisted method development was successfully applied for the separation of Fab (fragment antigen binding) and Fc (fragment crystallisable region) variants of cetuximab in both salt- and pH- gradient modes (16,17,18). In salt gradient mode, 10 min and 30-min gradients were performed on a 100âmm long standard bore column at pH = 5.6, 6.0, and 6.4 to build up the models. In the pH gradient mode, the experiments were performed with 10-min and 30-min gradients at temperatures of 25 °C and 55 °C. Finally, similar separation quality was achieved in the two modes and the analysis times were comparable.
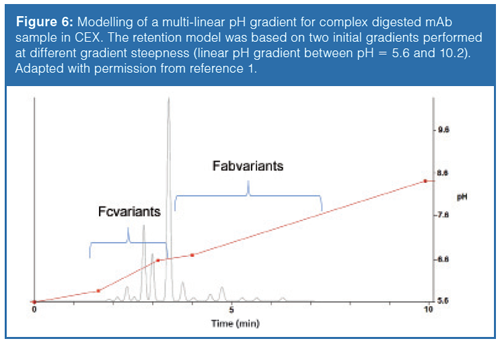
There are also some additional benefits of using modelling software, such as the possibility to simulate multilinear gradients. Multilinear gradients often improve the selectivity and can reduce analysis time. Figure 6 shows a calculated multilinear gradient separation for a complex papain digested mAb sample analyzed in pH gradient mode.

Hydrophobic Interaction Chromatography: Hydrophobic interaction chromatography (HIC) is able to separate protein species based on their hydrophobicity, under nondenaturing conditions. Currently, HIC is mostly applied for the characterization of mAbs, ADCs, and bispecific antibodies (bsAbs) (1). One of its most important applications is the separation of different populations of ADC molecules that differ in their number of drugs per antibody, which are often known as DAR (drugâtoâantibody ratio) species.
In HIC, the separation of proteins is based on a negative salt gradient performed on a slightly hydrophobic stationary phase, using physiological mobile phase pH conditions. The retention mechanism in HIC is often misunderstood and none of the proposed theories have received general acceptance. Retention in HIC mostly depends on the ligand type, ligand chain length, and ligand density. It has been shown that the salt type can have different effects on the retention, depending on the hydrophobicity of the protein to be separated. The influence of salts in HIC follows the lyotropic (Hoffmeister) series for the precipitation of proteins from aqueous solutions. In practice, sodium or ammonium sulfate effectively promote stationary phase–protein interactions and have a stabilizing effect on protein structure. Besides the salt type, salt concentration also influences retention in HIC. Depending on the lyotropic strength of the various salts, different concentrations are required to maintain the same saltingâout effect.
The most efficient method development in HIC is based on either (i) gradient steepness–temperature (tG × T) or on (ii) gradient steepness–organic modifier (tG × Corganic) models. The addition of organic modifier to the mobile phase can improve the recovery of the most hydrophobic species (for example, high DARs of ADCs) and can also impact their retention. Therefore, organic modifier content can also be a useful method variable when highly hydrophobic proteins have to be analyzed in HIC. Both protic and aprotic solvents can be used (for example, isopropanol, acetonitrile) since they affect the salting-out process in different ways through their different solvation. Recently, a linear HIC retention model has been derived for organic modifier content, and therefore the impact of organic modifier can be easily studied using only two levels (for example, 5 and 15%).
Custom-made retention models have been successfully applied for the separation of intact mAbs and ADC species for both “gradient steepness–temperature” and “gradient steepness–organic modifier” modes (19). For ADC species, a multilinear gradient (steeper at the beginning and flatter at the end) or logarithmic gradient gives better selectivity than linear gradients, probably because ADC species consist in a homologous series of linkers and cytotoxic drugs (20).
Conclusion
This work illustrates the possibilities offered by HPLC modelling software for the method development of small molecules as well as biopharmaceuticals. Such software can be used to effectively develop methods by the virtual screening of several analytical conditions (that is, gradient time and composition, pH, temperature, ternary mobile phase composition), based on a limited number of initial experimental runs. This allows the analyst to rapidly and efficiently develop a chromatographic method. Besides method development, robustness testing and method transfer between different instruments and column dimensions can also be virtually evaluated using HPLC modelling software.
Acknowledgements
The authors wish to thank Alain Beck from Pierre Fabre for providing the antibody–drug conjugate sample. Davy Guillarme wishes to thank the Swiss National Science Foundation for support through a fellowship to Szabolcs Fekete (31003A 159494).
References
- E. Tyteca, J.L. Veuthey, G. Desmet, D, Guillarme, and S. Fekete, Analyst141, 5488–5501 (2016).
- J.M. Davis and J.C. Giddings, Anal. Chem. 55, 418–424 (1983).
- P. Petersson, J. Manch, M.R. Euerby, A. Vazhentsev, M. McBrien, S.K. Bhal, and K. Kassam, Chromatogr. Today 15–18 (2014).
- E.F. Hewitt, P. Lukulay, and S. Galushko, J. Chromatogr. A1107, 79–87 (2006).
- R.M. Krisko, K. McLaughlin, M.J. Koenigbauer, and C.E. Lunte, J. Chromatogr. A 1122, 186–193 (2006).
- J.A. Lewis, L.R. Snyder, and J.W. Dolan, J. Chromatogr. A721, 15–29 (1996).
- I. Molnár, H.J. Rieger, and K.E. Monks, J. Chromatogr. A1217, 3193–3120 (2010).
- R. Kormány, I. Molnár, and J. Fekete, J. Pharm. Biomed. Anal. 135, 8–15 (2017).
- International Conference for Harmonization, ICH Q2(R1) Guideline (ICH, Geneva, Switzerland, 2005).
- B. Dejaegher and Y. Vander Heyden, J. Chromatogr. A 1158, 138–157 (2007).
- Y. Vander Heyden, A. Nijhuis, J. Smeyers-Verbeke, B.G.M. Vandeginste, and D.L. Massart, J. Pharm. Biomed. Anal.24, 723–753 (2001).
- R. Kormány, J. Fekete, D. Guillarme, and S. Fekete, J. Pharm. Biomed.Anal.89, 67–75 (2014).
- R. Kormány, J. Fekete, D. Guillarme, and S. Fekete, J. Pharm. Biomed. Anal. 94, 188–195 (2014).
- S. Fekete, S. Rudaz, J. Fekete, and D. Guillarme, J. Pharm. Biomoed. Anal.70, 158–168 (2012).
- S. Fekete, I. Molnár, and D. Guillarme, J. Pharm. Biomoed. Anal. 137, 60–69 (2017).
- G. Teshima, M.X. Li, R. Danishmand, C. Obi, R. To, C. Huang, J. Kung, V. Lahidji, J. Freeberg, L. Thorner, and M. Tomic, J. Chromatogr. A1218, 2091–2097 (2011).
- S. Fekete, A. Beck, J. Fekete, and D. Guillarme, J. Pharm. Biomed. Anal.102, 33–44 (2015).
- S. Fekete, A. Beck, J. Fekete, and D. Guillarme, J. Pharm. Biomed. Anal.102, 282–289 (2015).
- M.R. Rodriguez, D. Guillarme, A. Beck, and S. Fekete, J. Pharm. Biomed. Anal.118, 393–403 (2016).
- B. Bobály, G.M. Randazzo, S. Rudaz, D. Guillarme, and S. Fekete, J. Chromatogr. A1481, 82–91 (2017).
Szabolcs Fekete holds a Ph.D. in analytical chemistry from the Technical University of Budapest, Hungary. He worked at the Chemical Works of Gedeon Richter Plc at the analytical R&D department for 10 years. Since 2011, he has worked at the University of Geneva in Switzerland in the group of analytical pharmaceutical chemistry. He has contributed 90+ journal articles. His main interests include liquid chromatography, column technology, method development, pharmaceuticals, and protein analysis.
Róbert Kormány is a chemist and special analyst in chromatography. He graduated from the University of Debrecen then completed his education with an additional degree in chromatography and a Ph.D. in chemical sciences from the Department of Inorganic and Analytical Chemistry of the Budapest University of Technology and Economics in the laboratory of Prof. Dr. Jenö Fekete. He works at the pharmaceutical company Egis and specializes in developing UHPLC methods for the separation of pharmaceutical compounds using computer modelling.
Davy Guillarme holds a Ph.D. in analytical chemistry from the University of Lyon, France. He is now senior lecturer at the University of Geneva in Switzerland. He has authored 180 journal articles related to pharmaceutical analysis. His expertise includes HPLC, UHPLC, HILIC, LC–MS, SFC, and the analysis of proteins and mAbs. He is an editorial advisory board member of several journals including Journal of Chromatography A, Journal of Separation Science, and LCGC North America.







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.


, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

