Mitigating the Effect of Surfactants in Sample Preparation of a Phospholipid Formulation for Impurities Analysis by Reversed-Phase Liquid Chromatography
LCGC North America
A surfactant based diluent was used in sample preparation for reversed phase HPLC analysis of a drug product and its impurities in a phospholipid formulation. The use of the didodecyl trimethylammonium bromide (DDAB) enabled a consistent, quantitative extraction of the analytes of interest from the lipid matrix in a much more aqueous, weak solvent. Water was added as an anti-solvent to precipitate out the surfactant from the system to eliminate HPLC injection pressure spikes, enabling large volume injections and achieving a consistent, robust method for long term use. Method development and validation steps are described.
A surfactant-based diluent was used in sample preparation for reversed-phase high performance liquid chromatography (HPLC) analysis of a drug product and its impurities in a phospholipid formulation. The use of didodecyl trimethylammonium bromide (DDAB) enabled a consistent, quantitative extraction of the analytes of interest from the lipid matrix in a much more aqueous, weaker solvent than was specified in a previously established method. Water was added as an antisolvent to precipitate the surfactant from the system to eliminate HPLC injection pressure spikes, thereby enabling large-volume injections and achieving a consistent, robust method for long-term use. This article describes both the method development and validation steps.
Among the many applications of surfactants, or surface-active agents, in the chemical and pharmaceutical industry, their use in sample preparation in analytical operations play an important role. Many chromatographers focus on the separation of the analytes to achieve good resolution, peak shape, and sensitivity, not realizing that many problems can arise from preliminary sample preparation steps, which are often the weakest link of the analysis. The preference of most analysts for the analytical sample preparation is to dilute and inject the sample into the chromatograph, with additional clarification (extraction and separation) steps added only when necessary.
Surfactants have been used in sample preparations for both organic and inorganic compounds (1). They can play the role of an emulsifier, a micellar extraction medium, and an ion-pairing agent for extraction, or a more complicated role such as in surfactant-coated magnetic nanoparticle separations. Surfactants are also used for the determination of drugs in serum and urine by micellar liquid chromatography (LC) (2). In the quality control (QC) environment, surfactants are routinely used for dissolution testing, as an additive in the dissolution media. Most often used is sodium dodecyl sulfate (SDS), also known as sodium lauryl sulfate (SLS), for sparingly water-soluble drug products (3,4).
Recent developments in sample preparations employing surfactants for the analysis of pesticide residues in food and environmental samples by microextraction techniques include cloud-point extraction, dispersive liquid–liquid microextraction, and ultrasound-assisted surfactant-enhanced emulsification microextraction (5).
This article describes the development of a stability-indicating analytical method for a drug product in a phospholipid formulation for impurities analysis at low parts-per-million levels by reversed-phase high performance liquid chromatography (HPLC) using surfactants in sample preparation.
Background
We had to adapt a validated stability indicating, reversed-phase HPLC for determining impurities in a dry powder inhalation formulation, in early phase development, that comprised the phospholipid 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), calcium chloride, and the active pharmaceutical ingredient (API). The existing method had used a sample and standard diluent of 80:20 (v/v) methanol–water to extract the API and related impurities and degradants from the formulation matrices with 2.5–20% (w/w) drug load. For the 2.5% drug load formulation, the API and related impurities were quantitatively extracted from a 10-mg/mL semiviscous slurry of the formulation. After centrifugation of the slurry, the supernatant was injected onto the HPLC system for analysis.
A new formulation arose with the same excipients, but at only a 0.4% drug load. By regulatory guidance (6), impurities at greater than 0.1% (relative to active ingredient content) limit of quantitation (LOQ) in the drug product need to be quantitated and reported. When considering the overall formulation composition, the LOQ of 0.1% is equivalent to 4 ppm in our formulation that needs to be quantitated for each potential impurity that may arise during stability studies.
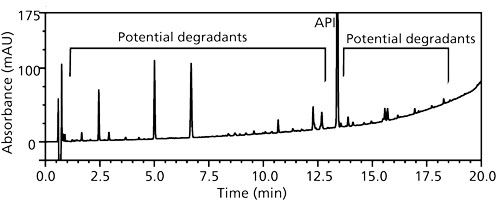
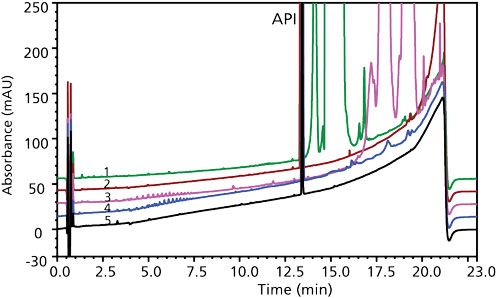
The chromatographic conditions were deemed suitable and did not need modification because forced degradation studies had demonstrated suitable separation of potential degradation products. The API was very stable in extreme high- and low-pH conditions and was susceptible to only photoinduced degradation (Figure 1).


Figure 1: Photodegradation of solid active pharmaceutical ingredient (API) showing potential degradants throughout the gradient elution window.
Experimental
HPLC Method
The gradient mobile phases consisted of 0.1% (v/v) trifluoroacetic acid in water as mobile-phase A and 0.1% (v/v) trifluoroacetic acid in 90:10 (v/v) acetonitrile–methanol as mobile-phase B. An Agilent 1260 HPLC system equipped with a diode-array absorbance detector was used for the analysis. The chromatography column was a 100 mm x 4.6 mm, 2.6-µm Phenomenex Kinetex C18 column operated at 45 °C. The detection wavelength was 230 nm. The injection volume of the original method was 10 µL, with a column loading of ~2.5 µg of the API. The gradient elution used a flow rate of 1.6 mL/min with the following program: 15–60% B (0–13.8 min), 80% B (18.4 min), 95% B (20.0 min), 15% B (20.1–23.0 min).
Sample Preparation Method Development
The method development strategy was to leverage the validated HPLC method chromatographic conditions and only adapt the sample preparation procedure to ensure quantitative extraction of the new formulation. To achieve the required sensitivity and LOQ, the column loading of ~2.5 µg of API from the original method needed to be maintained as closely as possible. This column loading was achieved by carefully selecting the extraction diluent, sample concentration, and injection volume. If the extracted API concentration must be lowered, for example, a proportional increase in injection volume could be used to compensate and maintain the same column loading. Alternatively, an extended-pathlength flow cell was also evaluated.
Results and Discussion
Evaluation Using an Extended-Pathlength Flow Cell
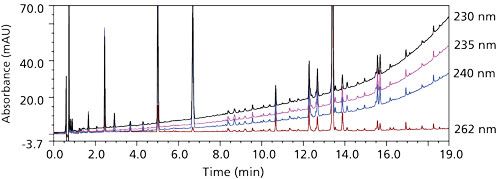
The HPLC system used was configured with a 6-cm pathlength flow cell instead of a typical 1-cm flow cell. In theory, the increased pathlength compensates for the sixfold drop in drug loading (from 2.5% to 0.4%) and corresponding drop in sample concentration without any loss in sensitivity or change in sample preparation procedure or injection volume. However, because of the relatively low detection wavelength (230 nm), in conjunction with use of methanol (10% in mobile phase B), which has an ultraviolet (UV) cutoff at 210 nm (7), a steep rise in the baseline was observed that might interfere with consistent integration of small impurity peaks. Some consideration was given to shift to a higher detection wavelength, away from the spectral maximum of the API. As the detection wavelength increased, the baseline drift resulting from the gradient would decrease. However, this result was not optimal, as there was loss in sensitivity of impurity peaks, such as the impurity at ~6.8 min in Figure 2 (compare trace at 262 nm to that at 240 nm).

Figure 2: Detection of forced degraded API at various wavelengths. Top to bottom: 230 nm, 235 nm, 240 nm, and 262 nm.
Assessment of Suitable Injection Volumes
The main purpose of a stability indicating impurities HPLC method in pharmaceutical development is to detect and quantitate degradants in the course of long-term ICH stability studies (8) of at least 1 year or more. The impurity profiles and relative ratios of degradants of a long-term stability study may not correspond to those observed under accelerated force degradation conditions (9–11). Therefore, the method needs to be able to detect all minor and major degradants observed in forced degradation. As mentioned above and shown in Figure 1, forced degradation of our API generated impurities that were eluted early and late, throughout the gradient elution window. Any compositional change to the sample diluent or injection volume to accommodate the analysis of the new 0.4% drug load formulation must not deleteriously affect the peak shape of these peaks of interest, particularly the early eluted impurities that are more susceptible to such changes. Therefore, a precursory study was conducted to identify the highest injection volumes tolerable (without causing peak splitting) with various ratios of methanol–water in the diluent, with or without basification with ammonium hydroxide. Basic pH in aqueous conditions was known to dramatically improve the solubility of the acidic API and potentially improve extraction efficiency.
It was important to conduct this injection volume study up-front because achieving the necessary column loading to meet the LOQ requirement could potentially violate conventional “rules of thumb” and may lead to poor peak shapes or split peaks in LC: Injection of sample in a solvent stronger than the initial gradient mobile phase composition, a large volume injection of such a sample, or mismatch of pH between the ionizable analytes in the sample injection and the mobile phase (12–14).
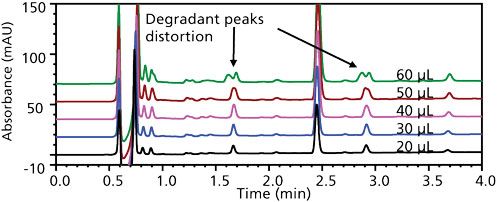
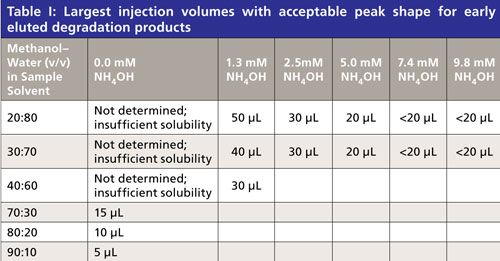
A photodegraded API sample was dissolved with various methanol–water ratios and various concentrations of ammonium hydroxide and injected with incrementally larger injection volumes up to 100 µL. The chromatograms were overlaid and visually assessed to determine the largest tolerable injection volume that did not cause apparent peak splitting of the earliest eluted significant degradant peaks. An example is given in Figure 3 demonstrating the early eluted peaks splitting at retention times of ~1.7 and ~2.9 min. The largest tolerable injection volumes without peak splitting, summarized in Table I, at any given composition of solvent strength (including basic modifier) dictated the sample concentration required to achieve the target sensitivity. Below 40% methanol, the solubility of the API was appreciably reduced and basification (high pH) was necessary to dissolve it.

Figure 3: Injection volumes (top to bottom): 60, 50, 40, 30, and 20 µL of photodegraded API dissolved in 30:70 (v/v) methanol–water with 1.3 mM ammonium hydroxide. In this example, 40 µL is considered the largest acceptable injection volume.

Sample Recovery Studies
Because the method was intended for use in a GMP QC laboratory, it needed to be kept as simple as possible to ensure consistent, reliable results. The original method, at the 2.5% drug load, called for extracting the sample with 80:20 (v/v) methanol–water, at a 10-mg/mL formulation sample concentration, using ultrasonication for agitation of the slurry solution. Following agitation, the semiviscous slurry suspension was centrifuged for sample clarification, and a 10-µL volume of the supernatant was injected, the maximum tolerated injection volume at 80% (v/v) methanol. With the new formulation at a 0.4% drug load, approximately one-sixth of the original formulation, a sixfold increase in the formulation sample concentration was attempted. However, because the phospholipid matrix was only wetted by the solvent at such a high concentration, the sample took on an appearance and consistency of dried white glue. It was obvious that a combination of increased injection volume accompanied with a sample concentration lower than 60 mg/mL was required to achieve the needed sensitivity.
A two-pronged strategy was considered: Optimize the methanol–water ratio in conjunction with diluent basification to improve the drug solubility, and use surfactants to partially dissolve and disrupt the phospholipid matrix to allow the diluent to access and extract the API and impurities.
Optimization of Methanol–Water Ratio with Diluent Basification
Samples prepared in the diluent with 70% or 80% methanol, at progressively lower formulation sample concentrations, down to 30 mg/mL, remained uninjectable and paste-like. At sample concentrations of 20 mg/mL and lower, the sample could be centrifuged and the supernatant recovered for injection. Reasonable recoveries (~100%) were obtained. However, at such sample concentrations and the limiting injection volume, the required sensitivity could not be achieved.
At a lower methanol content of 30–40% (v/v), which can tolerate a 3–4-fold increase of the injection volume, the sample required a slight basification; otherwise, the API essentially had no solubility as mentioned previously. However, the extraction of the API from the phospholipid matrix resulted in incomplete recovery (~50–85%) even at the lower formulation concentration ranges of 8–20 mg/mL. Efforts with different orders of addition of basified 100% organic or 100% aqueous solvent also failed to achieve full extraction of the API from the formulation.
Use of Surfactants
The three general categories of surfactants-anionic, neutral, and cationic-were screened for their effectiveness to extract the API. The surfactant, as an additive to a high sensitivity impurities solvent extraction method, needed to meet some stringent requirements: The surfactant needs to have solubility in water or water–methanol mixture, since the target solvent system needs to be highly aqueous (to allow for high injection volume); the surfactant needs to be chemically inert to the API and related impurities to avoid generation of false positive impurity peaks; and the surfactant needs to be sufficiently pure that it does not contain impurities of its own that would interfere with potential API impurities quantitation.
Anionic Surfactant
Only one anionic surfactant of this category, sodium lauryl sulfate, was tried, because there was practically no wetting of the formulation powder with this surfactant.
Nonionic Surfactants
The nonionic surfactants studied (Table II) were all ethylene oxide–based surfactants that yielded essentially full extraction of the drug. A majority of the commercially available surfactants evaluated contained a high number of potentially interfering peaks. These peaks presumably were either the surfactant itself or shorter chain polymer impurities. This result was not unexpected with a surfactant such as Triton X-100 that contains a strong chromophore. However, even surfactants structurally expected to be UV transparent (Tween 20, Tween 80, and Brij 35) exhibited significant peaks that might interfere with potential API degradants (Figure 4). One surfactant, polidocanol, did hold some promise, as it yielded an interference-free baseline. Further optimization was conducted with a 2x2 experiment design study with methanol ranging from 30–40% and polidocanol ranging from 0.5–1.5% (w/v). The results (Figure 5) showed that a diluent containing 1.5% (w/v) polidocanol and 30% (v/v) methanol was a good candidate sample solvent. Working with 30% (v/v) methanol allowed for a 40-µL injection volume (Table I). When accompanied with a ~16-mg/mL sample concentration (~64 µg/mL API), the target column loading of the extracted API of ~2.5 µg could be achieved. Polidocanol also enhanced the solubility of the API to the extent that basification of the diluent was not required. However, the prepared sample solution generated additional solution degradation impurity peaks (data not shown). The presence of residual reactive peroxides from the synthesis of the ethylene oxide polymers was suspected to cause these additional peaks. Addition of an antioxidant (thiourea) mitigated the extra peaks, but when this approach was used with a forced-degradation sample containing API-related potential degradants, it imparted changes to the impurity profile. This result was not acceptable because it has the potential to cause inaccurate quantitation of the impurities.

Figure 4: Chromatograms of samples extracted with methanol–water diluents containing nonionic surfactants: 1 = Triton X-100, 2 = Brij 23, 3 = Tween 80, 4 = Tween 20, 5 = polidocanol.

Cationic Surfactants
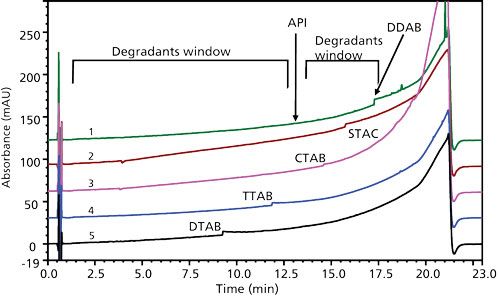
A series of quaternary ammonium salts with trimethyl groups and varied alkyl chain lengths (Table II) was initially assessed at a 50 mM concentration in a solvent with 40:60 (v/v) methanol–water, with and without basification. In addition, several commercially available quaternary ammonium salts containing two, three, or four long-chained alkyl groups were evaluated in the same manner. All of the trimethyl quaternary ammonium and dimethyl quaternary salts did yield essentially quantitative API extraction and reasonably clean baselines. However, each progressively longer alkyl chainlength surfactant diluent exhibited a steep baseline step shift at progressively longer retention times (Figure 6). The “step” in each chromatogram was actually each surfactant being eluted as a poorly shaped, mass overloaded tailing peak with very low UV absorbance signal. The majority of these surfactants could not be used because they were eluted at a retention time that could interfere with the quantitation of potential degradation products, except for didodecyldimethylammonium bromide (DDAB). Because of DDAB’s higher hydrophobicity with two C12 alkyl chains, it was retained the longest of the surfactants studied and was eluted in a region, after the vast majority of the potential degradants were eluted, that minimized any potential analyte interferences.

Figure 6: Chromatograms of cationic quaternary ammonium surfactants displaying baseline steps: 1 = DDAB, 2 = STAC, 3 = CTAB, 4 = TTAB, 5 = DTAB.
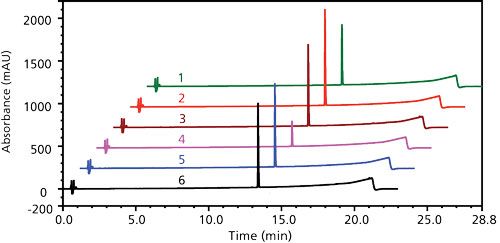
The target sample concentration needed to be ~10–20 mg/mL of formulation, which corresponded to ~12–24 mM of DSPC. Likely the 50 mM concentration of DDAB used in screening was in excess. Decreasing the DDAB concentration was also desirable to reduce the height of the apparent baseline step. The DDAB peak would also be less overloaded, increasing its apparent retention time and moving it further away from degradant peaks. Screening in the range of 2–10 mM DDAB, in 30:70 (v/v) methanol–water, 5 mM was identified as the minimum DDAB concentration to obtain full extraction of the API. At 2 mM DDAB, there was a precipitous drop in the API extraction, but the addition of 1 mM of ammonium hydroxide for diluent basification yielded markedly higher extraction (Figure 7). In addition to full recovery, the extracted sample’s impurity profile remained unchanged. Thus, suitable sample solution stability was also achieved.

Figure 7: Sample extractions screening different concentrations of DDAB, with or without 1 mM ammonium hydroxide, all in 40:60 (v/v) methanol–water: 1 = 2.5 mM DDAB/1 mM NH4OH, 2 = 5 mM DDAB/1 mM NH4OH, 3 = 10 mM DDAB/1 mM NH4OH, 4 = 2.5 mM DDAB/no NH4OH, 5 = 5 mM DDAB/no NH4OH, 6 = 10 mM DDAB/no NH4OH.
However, during the DDAB concentration optimization, sample injection-induced pressure spikes were observed that did not correlate with DDAB concentration, the only variable being changed in a series of injections shown in Figure 8. Although the HPLC system could tolerate such pressure spikes (up to 600 bar) and the back pressure did not exhibit any hysteresis, these random pressure spikes were undesirable because the potential existed to exceed the system pressure limit, which could cause injection sequences to be interrupted on long-term use and posed a higher risk for column damage. Additional high-speed centrifugation (15,000 rpm) to remove undissolved solids before injection failed to eliminate the pressure spikes.

Figure 8: Pressure profiles of injected samples prepared with different concentrations of DDAB in the diluent: 1 = 4 mM DDAB, 2 = 8 mm DDAB, 3 = 6 mM DDAB, 4 = 10 mM DDAB.
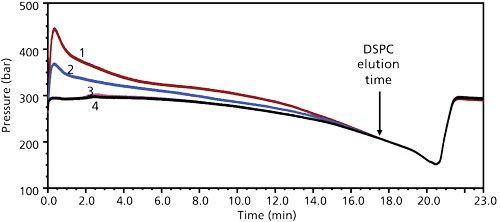
DDAB met all the criteria needed for use in this method (solubility in methanol–water, full API and impurities extraction, chemical inertness), except for the undesirable injection pressure spikes. DDAB may still be used, if somehow it can be removed from the system after API extraction. With removal of DDAB, the dispersability of the suspended solids perhaps would be reduced, enabling better phase separation in the centrifugation clarification step. Initial attempts to add inorganic salts (KCl and Na2SO4) as counterions to precipitate the quaternary ammonium cations were not successful. Addition of SDS after DDAB–methanol–water–base extraction did bring a distinct phase separation after centrifugation. Unfortunately, the SDS addition also precipitated the API and led to low recovery of the API.
The solution to the pressures spikes turned out to be water addition. When DDAB was initially screened as the wetting and solubility agent, it was prepared in 30:70 (v/v) methanol–water. When we attempted to dissolve DDAB directly in 20:80 (v/v) methanol–water to prepare the diluent, the DDAB was difficult to dissolve and produced a very hazy solution. A brief solubility study identified that ~30% methanol was the minimum required to have appreciable dissolution of DDAB. Water behaved as an antisolvent with respect to DDAB solubility. Thus, the following sample preparation scheme was devised: The drug product at 20 mg/mL could first be extracted in 40:60 (v/v) methanol–water, containing 10 mM DDAB and 1.3 mM ammonium hydroxide for basification. (Higher drug product concentrations were found to be too viscous for efficient mixing and caused low recovery.) After extraction, an equal volume of water containing 1.3 mM ammonium hydroxide was added to induce precipitation of DDAB and reduce the dispersability of the system. The ammonium hydroxide base concentration was added to maintain sufficient solubility of the API. Upon centrifugation to bring about partial phase separation, the still semiturbid supernatant injected for analysis no longer had the pressure spike issue (Figure 9).

Figure 9: Pressure profiles of 35 consecutive injections of standard or samples in final diluent.
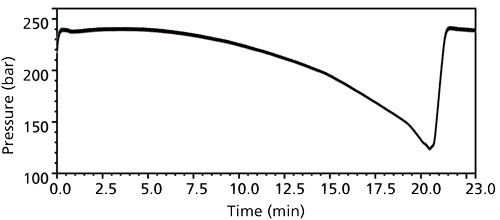
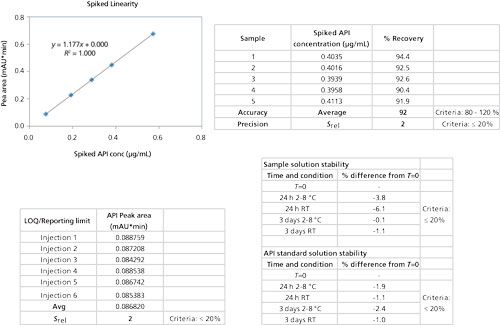
The final methanol-to-water ratio of 20:80 (v/v) in the sample allowed for injection volumes as high as 50 µL without causing peak splitting of potential early eluted peaks. The final injected API drug load was 2 µg, 80% of the original method’s column load. The slightly lower column load still gave sufficient sensitivity. Final method validation demonstrated consistent average spiked recovery of 92% at the impurity specification level, a relative standard deviation of 2%, and linearity R2 of 1.000 (Figure 10).

Figure 10: Summary of method validation results.
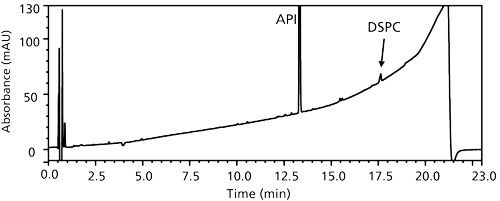
Why did such pressure spikes exist? The sample preparation was an extraction process to solubilize the API and related impurities into the bulk solution phase. The DDAB–methanol–water–base solvent system only partially dissolved the DSPC–calcium chloride matrix. The DDAB as a surfactant not only allowed wetting and partial dissolution of the highly hydrophobic excipients, but also acted as an effective dispersant of the phospholipid. Upon injection and mixing with the initial gradient mobile phase, a supersaturated condition for the phospholipid was created and induced precipitation that partially blocked the column. At approximately 18 min (Figure 11), when the gradient organic composition was sufficiently high to dissolve and elute the phospholipid (DSPC), the partial blockage was removed and the end gradient pressure profile behaved normally with no pressure hysteresis.
Figure 11: A representative sample chromatogram of final method.
In sample preparation for reversed-phase HPLC analysis, a sample is often dissolved in a diluent consisting of a mixture of water and an organic solvent. It is preferable to use a sample diluent that matches the mobile-phase composition of an isocratic method or the initial mobile-phase composition of a gradient method. Alternatively, it is also preferable to use a diluent that is weaker in eluent strength than the mobile-phase composition, because this approach provides at least two advantages in loading the sample to the HPLC column: It allows for sample peak compression, and it allows larger volume injection without peak splitting, particularly for early eluted peaks. The use of surfactants allowed us to take advantage of these benefits to achieve the method goals.
Conclusion
We started from a pharmaceutical phospholipid sample preparation procedure that used a mostly organic (80% methanol) sample extractant for reversed-phase LC analysis. With constraints in injection volume and sample solubility, it was initially not obvious how to maintain the sensitivity of the method with a sixfold decrease in API concentration caused by the formulation changes from 2.5% to 0.4% drug load. The use of the appropriate cationic surfactant enabled a consistent, quantitative extraction in a much more aqueous, weaker solvent. Water was added to the sample, as the weak solvent, to enable a large-volume injection and as an antisolvent to remove the surfactant (effective dispersant) from the system to eliminate pressure spikes and achieve a consistent, robust method for long-term use in the QC laboratory.
References
- M. Moradi and Y. Yamini, J. Sep. Sci. 35, 2319–2340 (2012).
- J. Esteve-Romero, J. Albiol-Chiva, and J. Peris-Vicente, Anal. Chim. Acta926, 1–16 (2016).
- U.S. Pharmacopeia-National Formulary [USP 39 NF 34], Volume 1. Rockville, Maryland: United States Pharmacopeial Convention, Inc; 2015. [711] Dissolution; p. 540–551, 2016.
- C. Noory, N. Tran, L, Ouderkirk, and V. Shah, Dissolution Technol. 7(1), 16–18 (2000).
- Y. Santaladchaiyakit, S. Srijaranai, and R. Burakham, J. Sep. Sci. 35, 2373–2389 (2012).
- ICH Harmonized Tripartite Guideline - Impurities in New Drug Product Q3B(R2), June 2006.
- W.M. Haynes, Ed., CRC Handbook of Chemistry and Physics, 96th edition (Internet version 2016, CRC Press/Taylor and Francis, Boca Raton, Florida, 2016).
- ICH Harmonized Tripartite Guideline - Stability Testing of New Drug Substances and Products, 2003.
- J. Kogawa and H. Salgado, Curr. Pharm. Anal. 12, 18–24 (2016).
- M. Blessy, R. Patel, P. Prajapati, and Y. Agrawal, J. Pharm. Anal. 4(3), 159–165 (2014).
- S. Singh, M. Junwal, G. Modhe, H. Tiwari, M. Kurmi, N. Parashar, and P. Sidduri, Trends Anal. Chem. 49, 71–88 (2013).
- C. Hawkins and J. Dolan, LCGC North Am. 21(12), 1134–1138 (2003).
- J. Dolan, LCGC North Am. 23(3), 266–271 (2005).
- M. Dong, Modern HPLC for Practicing Scientists (J. Wiley & Sons, Hoboken, New Jersey, 2016).
James Tam and Andrei Blasko are with Novartis Pharmaceuticals Corporation in San Carlos, California. Please direct correspondence to: james.tam@novartis.com



















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



