Micellar Liquid Chromatography: How to Start
LCGC Europe
Micellar liquid chromatography (MLC) is a reversed-phase liquid chromatographic mode with a solution of surfactant forming micelles as the mobile phase. The interaction of solutes with the stationary phase coated with surfactant monomers, combined with the increased solubilization capability of micelles, have profound implications with regard to retention, selectivity and efficiency. Practical steps that a chromatographer involved in MLC should consider when developing an analytical procedure are described, including mobile phase preparation, column conditioning and cleaning.
Micellar liquid chromatography (MLC) is a reversed-phase liquid chromatography (RPLC) mode with a solution of surfactant, containing either ionic or non-ionic head groups at concentrations exceeding the critical micellar concentration (cmc), as the mobile phase.1–4 The mobile phase contains, therefore, micelles in addition to surfactant monomers, whereas the stationary phase is coated by adsorption of surfactant monomers, forming a surface similar to the exterior of micelles. The existence of micelles in the mobile phase and the modification of the stationary phase surface affects retention, selectivity and efficiency.
Non-polar solutes eluted with mobile phases of either non-ionic or ionic surfactants and ionizable solutes eluted with mobile phases of non-ionic surfactants will only experience non-polar, dipole–dipole and proton donor-acceptor interactions with micelles and stationary phase.5 Ionizable solutes will also interact electrostatically with the charged outer-layer of ionic micelles and the charged surfactant layer on the stationary phase. In any instance, the steric factor can also be important. Solutes are separated on the basis of their differential partitioning between bulk aqueous phase and micelles in the mobile phase, and between bulk aqueous phase and surfactant-coated stationary phase.



Figure 1
MLC makes use of the same hardware (pumps, injectors, tubing, detectors, etc.) as classical RPLC with organic solvent–water mixtures. The most interesting characteristics offered by MLC with regard to classical RPLC are its large versatility produced by the different kinds of solute-micelle and solute-modified stationary phase interactions, the change in selectivity, the suppression of peak tailing for basic drugs chromatographed with conventional columns, the analysis of samples containing compounds in a wide range of polarities using isocratic elution, and the direct injection of physiological fluids, avoiding the tedious sample pre-treatment required in classical RPLC (see Figures 1–3). The application field of MLC is summarized in Table 1.


Figure 2
Despite these advantages and others that will be commented on in this report, the applicability of the technique in the analytical laboratories has been limited. This could partially be attributed to the main drawbacks described in the early reports of this technique: the weak eluting power and reduced efficiency of pure micellar mobile phases. Some of the solutions proposed along the years to overcome these limitations, such as the addition of small amounts of alcohol,11 and more recently, the use of large pore stationary phases,12 have improved the potential of the technique, but have not extended its use. The only real limitation of this chromatographic mode up to-date is related to the use of mass spectrometric detection, because direct on-line coupling to MLC is hindered by the presence of high concentrations of surfactant in the mobile phase.


Figure 3
After almost three decades of MLC experience with a great volume of scientific production, mainly related to the analysis of drugs in pharmaceuticals and physiological fluids (Table 1), the descriptions on the protocols to follow in the MLC routine are insufficient or unclear. In this report, we explain practical steps that a chromatographer involved in the MLC work should consider when developing an analytical procedure. The importance of column conditioning and cleaning is highlighted.


Table 1: Application field of MLC: reported procedures.a
Mobile Phase Preparation
Although pure micellar mobile phases are sometimes used, most separations in MLC are performed with hybrid micellar mobile phases in a buffered medium, that is, micellar solutions containing a small amount of an organic solvent, mainly a short-chain alcohol, with propanol being the most common. The proper stability of the micellar mobile phase is essential.
Critical micellar concentration: A suitable surfactant for MLC should have a low cmc. A high cmc would imply operating at high surfactant concentration, which would result in viscous solutions, giving undesirable high system pressure and background noise in UV detectors. The selection is often limited to the following surfactants: the anionic sodium dodecyl sulphate (SDS), the cationic cetyltrimethylammonium bromide (CTAB) and the non ionic Brij-35, whose main characteristics are summarized in Table 2. The cmc values of these surfactants in pure water are low enough for MLC. It should also be taken into account that the cmc is strongly affected by the presence of an organic solvent (Figure 4). The changes are related to the modification of the structure of the micelle, which also induces, at least partially, the reduced retention in MLC.15


Table 2: Characteristics of the most common surfactants in MLCa
Krafft point: The Krafft point is defined for ionic surfactants as the temperature at which the solubility of a surfactant monomer becomes equal to the cmc.16 Below the Krafft point temperature, the solubility is quite low and the solution appears to contain no micelles. Chromatographic work in MLC should be conducted above this temperature to avoid surfactant precipitation. This means that the Krafft point should be well below room temperature. Furthermore, to avoid ruining the column, laboratory temperature should always be above this value. This is especially critical in cold-climate regions where working in an air-conditioned laboratory will be mandatory. The Krafft point for SDS and CTAB is around 15 °C and 20–25 °C, respectively,17,18 but this is affected by the presence of salts. Thus, for SDS, it increases up to 18 °C in 0.1 M NaCl.18


Figure 4
Non-ionic surfactants also have a specific temperature, that if exceeded phase separation occurs, which is called the cloud point.14,19 Chromatographic work with these surfactants should be conducted below this temperature. This doesn't seem to be a problem, because the cloud point for the most common non-ionic surfactant in MLC, Brij-35, is ca. 100 °C for aqueous 1–6% solutions, whereas for Triton X–100 this value is 64 °C.14
Mobile phase pH: MLC employs the same packing materials as classical RPLC, which for conventional columns have a limited working pH range of 2.5–7.5. Appropriate pH values depend on the nature of the analytes and the surfactant selected. For instance, the separation of weak acids, using the anionic SDS, often requires pH fixed at 2.5–3, where the more retained protonated neutral species dominates.20 In these conditions, the separation space is wider and favours resolution. The pH of the micellar mobile phase is commonly fixed with phosphoric or citric acid buffers.1,4 For mobile phases containing SDS, potassium salts are not recommended as potassium dodecyl sulphate presents a high Krafft point and precipitates from aqueous solutions at room temperature.1 In any instance, the column should be equilibrated by purging with the mobile phase until the pH before and after the column is identical.
Type and concentration of organic solvent modifier: The selection of the appropriate organic solvent modifier in MLC should consider the polarities of the analytes. For polar compounds, sufficiently short retention times (below 20 min) are obtained with 1-propanol, 2-propanol or acetonitrile. For non-polar compounds or compounds with high affinity for the surfactant adsorbed on the stationary phase, stronger solvents as 1-butanol or 1-pentanol are needed.7 However, it should be noted that the two latter alcohols give rise to microemulsion formation at sufficiently high concentration.21
In practice, the amount of organic solvent that can be added is limited by its solubility. Additives that would normally not be considered in classical hydro-organic RPLC because of insufficient solubility can give rise to stable mobile phases with micelles. Thus, for instance, in 0.285 M SDS at 25 °C, the molar concentrations of 2-methyl-1-butanol, 1-pentanol, 1-hexanol and pentane were found to be 0.46, 0.92, 0.79 and 0.095, respectively, whereas their molar solubilities in water are only 6.1×10–3, 4.5×10–3, 1.2×10–3, and 9.5×10–6, respectively.22
Whereas, at high organic solvent concentration, the micelles disaggregate and the mobile phase contains only free surfactant molecules. The organic solvent contents that preserve the integrity of micelles are below 15% for propanol and acetonitrile, 10% for butanol and 6% for pentanol.22 These contents are low in comparison with those needed in classical reversed-phase liquid chromatography (RPLC). The lower organic solvent consumption results in reduced cost and toxicity, which may become prominent for "green chemistry". Also, the stabilization of the organic solvent in the micellar media decreases the risk of evaporation. This means that micellar mobile phases can be preserved in the laboratory for a long time without significant changes in their composition.
Surfactant Column Coating
The alkyl-bonded C18 is the stationary phase most widely used in MLC, but other columns can be selected (e.g., C8 and cyanopropyl). Alkyl-bonded phase columns are strongly modified when SDS, CTAB or Brij-35 are incorporated into the mobile phase. Adsorption isotherms of stationary phases of different nature in contact with mobile phases containing these three surfactants have been extensively studied, together with the effect of mobile phase additives on surfactant adsorption.13,23 The results provided information about column conditioning in MLC, which is useful to achieve reproducible results.
Mobile phase saturation: Pure and hybrid micellar solutions contain high amounts of water (usually more than 90% v/v) and are able to dissolve small amounts of silica, which could produce serious column damage. This is especially critical at >30 °C and/or pH > 6. For this reason, a saturating short column packed with 10 μm bare silica, or alternatively, the same packing as the analytical column, should be placed after the pump and before the injection valve to reduce pressure build-up.
Column conditioning: A column for MLC is generally stored in 100% methanol. Before starting column conditioning, the solvent should be replaced by 100% water. For this operation, a low
flow-rate (≤0.5 mL/min) should be selected at the beginning because of the high viscosity of the methanol–water mixture. Once the pressure decreases, the flow-rate may be raised. At least 30 column volumes of water are required to assure complete organic solvent removing. Now, the system is ready to be flushed with the micellar mobile phase. Different studies of column coating through surfactant breakthrough patterns have revealed that most surfactant adsorbs in less than one hour on the bonded-stationary phase.13,23 However, some additional surfactant continues to adsorb onto the column, especially in the instance of the non-ionic Brij-35.14
Mobile phase flushing: The micellar mobile phase should be continuously flushed through the system. If the chromatographic system is stopped during several hours, the micellar solution should not stay in contact with the bonded-silica based stationary phase to avoid surfactant precipitation. A static micellar mobile phase can also produce crystals around the pump plungers and seals. Such crystals may obstruct the system producing plugged connecting tubing and frits, seal failure, or scratched pistons.
A micellar mobile phase can be kept inside the chromatographic system overnight if the pump is not off. This avoids daily cleaning and re-equilibration. To reduce the cost, the mobile phase can be recycled, reducing the flow-rate to a minimal value (often 0.1–0.25 mL/min). However, it should be noted that in case of energy supply failure, column damage can occur. Mobile phase recycling is possible because of the low evaporation risk of organic solvents in hybrid micellar eluents. For the same reason, the micellar mobile phase can be recycled during the analysis, as long as a low number of injections is made.
Regeneration of surfactant coated reversed-phase columns: There is some concern about surfactant desorption from the column. Some reports claim that removing the surfactant-adsorbed layer for further use of a mobile phase of different nature (i.e., hydro-organic or containing another surfactant) is not possible. Thus, a small amount of SDS was reported to remain adsorbed on a Hypersil ODS column after cleaning,24 but other authors found that the surfactant layer was completely removed.13,25–27 This is the reason of the recommendation of keeping a column for the exclusive use of a given surfactant.
In general, regeneration can be appropriately performed with methanol, where most surfactants are highly soluble.27 The cleaning protocol comprises a two step procedure that takes about half an hour:
(i) First, the micellar mobile phase should be replaced by 100% pure water, by rinsing the chromatographic system with 10 to 20 column volumes of pure water. This step is necessary to avoid salt crystallization provoked by a brutal change from a buffered micellar mobile phase to 100% methanol.
(ii) Next, water will be replaced by 100% methanol to remove the adsorbed surfactant on the stationary phase. The same caution commented under "column conditioning" about the initial use of a low flow-rate should be followed. To assure complete surfactant desorption, at least 10 column volumes of methanol should be passed through the column.
A third final step can be included to assure complete recovery of the initial stationary phase surface. This consists in checking the retention times of a solute mixture (selected arbitrarily) with a hydro-organic mobile phase before and after using the micellar mobile phase. This step is not needed if the rules about column conditioning and cleaning are respected. A reduction in the lifetime of the columns used in MLC with respect to RPLC has not been observed. In fact, from our experience, column performance can be maintained for more than two years of intensive MLC use. Eventually, a small decrease in column efficiency may be attributed to column ageing.
Method Development
Therapeutic and doping control of drugs is one of the most important applications in MLC.9 Table 3 shows some examples on the use of MLC in this field. Hydrophobic interactions are scarcely affected by changes in the composition of micellar eluents. This implies that solutes of different hydrophobicity can be eluted in retention time windows narrower than in classical RPLC under isocratic elution (Figures 1 and 2). In case gradient elution is needed, equilibration times are also shorter in MLC.37,38

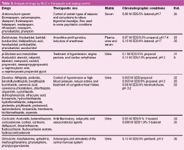
Table 3: Analysis of drugs by MLC in therapeutic and doping control
Interestingly, micellar media have been demonstrated to slow down the photochemical degradation of diverse drugs under the influence of UV radiation. The conditions that favour photodegradation processes can also be different in both aqueous organic and micellar media. For example, photochemical degradation of the diuretic furosemide at acidic pH is faster in hydro-organic media, but slower in a micellar solution of SDS.39 Stability conditions in micellar solutions should be, thus, checked before applying any MLC procedure to avoid undetected compounds.
One of the most interesting features of MLC is the possibility of direct injection of physiological fluids (e.g., urine, plasma, serum and milk) without any other pre-treatment than filtration, with no increase in system pressure or no noticeable damage after repetitive serial injections.10 The proteins, rather than precipitating on the column, are swept away harmlessly, eluting with or shortly after the solvent front. This could affect the detection of low retained compounds. However, it should be noted that SDS is the only surfactant usually used in MLC that efficiently solubilizes proteins in physiological matrices. Also, it is a good practice to prevent any possible column contamination by diluting or filtering the samples before injection. Some samples, such as serum, need centrifugation before chromatographic analysis. This little pre-treatment minimizes column exposure to undesirable compounds.
Acknowledgments
This work was supported by Projects CTQ2004-02760/BQU and CTQ2007-61828/BQU (Ministerio de Educación y Ciencia of Spain, MEC) and FEDER funds. M.J.R.A. thanks the MEC for a Ramón y Cajal contract.
María José Ruiz-Ángel obtained her PhD from the University of Valencia in 2003 under the direction of Dr M.C. García-Álvarez-Coque. Between 2004 and 2006, she was granted with a post-doctorate fellowship at the Laboratoire des Sciences Analytiques of the University Claude Bernard in Lyon (France) under the supervision of Dr A. Berthod, Since January 2007, she has been a senior researcher (Ramón y Cajal position) in the University of Valencia (Spain).
María Celia García-Álvarez-Coque has been a full professor in analytical chemistry at the University of Valencia (Spain) since 1997. She has written 200 research articles, focusing on HPLC in the last 15 years, particularly on fundamental studies and the development of chemometrical methods to extract the potential information contained in chromatographic signals. She is a coauthor of the books Micellar Liquid Chromatography and Chemometrics.
References
1. A. Berthod and M.C. García-Álvarez-Coque, Micellar Liquid Chromatography, J. Cazes, Ed. (Marcel Dekker, New York, USA 2000).
2. M.C. García-Álvarez-Coque and J.R. Torres-Lapasió, Hybrid micellar mobile phases in Encyclopedia of Chromatography, J. Cazes, Ed. (Marcel Dekker, New York, USA 790–796, 2002).
3. M.C. García-Álvarez-Coque and J.R. Torres-Lapasió, Micellar liquid chromatography in Encyclopedia of Analytical Science, P.J. Worsfold, A. Townshend and C.F. Poole, Eds. (Elsevier, Oxford, Vol. 5, 164–172, 2005).
4. M.J. Ruiz-Ángel, M.C. García-Álvarez-Coque and A. Berthod, Separ. Purif. Rev., (2008, in press).
5. M.L. Marina et al., Microchem. J., 53, 215–224 (1996).
6. M.J. Ruiz-Ángel et al., Anal. Chim. Acta, 454, 109–123 (2002).
7. R.D. Caballero et al., J. Chromatogr. A, 947, 31–45 (2002).
8. M.J. Ruiz-Ángel et al., J. Chromatogr. B, 767, 277–283 (2002).
9. J. Esteve-Romero et al., Trends Anal. Chem., 24, 75–91 (2005).
10. M.C. García-Álvarez-Coque and S. Carda-Broch, J. Chromatogr. B, 736, 1–18 (1999).
11. J.G. Dorsey, M.T. DeEchegaray and J.S. Landy, Anal. Chem., 55, 924–928 (1983).
12. T.J. McCormick et al., Anal. Chem., 72, 294–301 (2000).
13. A. Berthod, I. Girard and C. Gonnet, Anal. Chem., 58, 1356–1358 (1986).
14. M.F. Bogerding and W.L. Hinze, Anal. Chem., 57, 2183–2190 (1985).
15. S. López-Grío, J.J. Baeza-Baeza and M.C. García-Álvarez-Coque, Chromatographia, 48, 655–663 (1998).
16. R.C. Murray and G.S. Hartley, Trans. Faraday Soc., 31, 183–189 (1935).
17. P. Sehgal and D.E. Otzen, Prot. Sci., 15, 890–899 (2006).
18. K. Beyer, D. Leine and A. Blume, Coll. Surf. B: Biointerf., 49, 31–39 (2006).
19. B. Delgado et al., Anal. Chim. Acta, 518, 165–172 (2004).
20. S. Carda-Broch et al., J. Chromatogr. A, 893, 321–337 (2000).
21. S.M. Briant and K.D. Altria, J. Sep. Sci., 27, 1498–1502 (2004).
22. S. López-Grío et al., Anal. Chem., 72, 4826–4835 (2000).
23. A. Berthod, I. Girard and C. Gonnet, Anal. Chem., 58, 1362–1367 (1986).
24. J.H. Knox and R.A. Hartwick, J. Chromatogr., 204, 3–21 (1981).
25. A. Berthod, I. Girard and C. Gonnet. "Stationary phases in MLC surfactant adsorption and interaction with ionic solutes" in Ordered Media in Chemical separations, W.L. Hinze and D.W. Armstrong, Eds. (ACS Symp. Ser.), American Chemical Society, Washington D.C., USA, 346, 130 (1987).
26. D.E. Keller, R.G. Carbonell and P.K. Kilpatrick, J. Colloid Interface Sci., 155, 124–136 (1993).
27. A. Berthod and A. Roussel, J. Chromatogr., 449, 349–360 (1988).
28. M.E. Capella-Peiró et al., J. Chromatogr. B, 780, 241–249 (2002).
29. Y. Martín-Biosca et al., J. Pharm. Biomed. Anal., 21, 331–338 (1999).
30. M.E. Capella-Peiró et al., Anal. Biochem., 309, 261–268 (2002).
31. I. Rapado-Martínez et al., Anal. Chem., 71, 319–326 (1999).
32. S. Carda-Broch, J.S. Esteve-Romero and M.C. García-Álvarez-Coque, Anal. Chim. Acta, 375, 143–154 (1998).
33. M.A. Rosado et al., J. Chromatogr. B, 748, 415–424 (2000).
34. Z.L. Chen and S.F. Wang, Anal. Lett., 30, 2315–2325 (1997).
35. A. Santos-Montes and R. Izquierdo-Hornillos, J. Chromatogr B, 724, 53–63 (1999).
36. M. Gil-Agustí et al., Chromatographia, 57, 51–57 (2003).
37. L.S. Madamba-Tan, J.K. Strasters and M.G. Khaledi, J. Chromatogr. A, 683, 321–334 (1994).
38. L.S. Madamba-Tan, J.K. Strasters and M.G. Khaledi, J. Chromatogr. A, 683, 335–345 (1994).
39. S. Carda-Broch et al., Analyst, 127, 29–34 (2002).




















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.

