Method from Mars? Coping with Chromatographic Legacies
LCGC North America


Legacy LC methods seem like they come from a different planet. This month, we look at which conditions to keep, and which ones to let go.
In method development for liquid chromatography (LC), instead of starting from scratch, there are a variety of possible starting points that we can use to save time. For example, we can look to column and instrument vendor application notes, the peer-reviewed literature, or perhaps methods developed for similar applications by coworkers within our organizations. One particularly common starting point is to use a method developed with objectives similar to our own, and that has been published by the United States Pharmacopeia (USP). Given the long history of some of these methods, however, simply adopting older methods wholesale may involve the implementation of conditions that not only appear very strange to younger chromatographers, but have been rendered practically obsolete, given advances in LC technologies over the past few decades. In some highly regulated environments, the user does not have much flexibility to make changes to the established method, even if it is obviously outdated. However, in recent years there has been an increase in flexibility afforded to method developers to adapt to changes in LC technology by widening the scope of allowable changes to existing methods. Making sound decisions about which elements of an older method should be carried forward in the development of a new method requires a solid understanding of why particular conditions were used in the past, and whether or not they are still needed in light of new developments in LC column and instrument technologies. For this installment of "LC Troubleshooting" I have asked Tony Taylor of Crawford Scientific to join me in discussing some of the chromatographic legacies he encounters in his day-to-day work in his laboratories and those of his clients.
Dwight Stoll
Triethylamine as a Mobile Phase Additive: Take It or Leave It?
It is very common to find reversed-phase LC methods, most often designed for the analysis of amine-containing bases (for example, benzylamine), involving the use of triethylamine as a mobile-phase additive in the USP compendium, in vendor application notes, and in the peer- reviewed literature. For example, the USP methods for irbesartan, donepezil, and lamotrigine all call for triethylamine in the mobile phase (1). On the other hand, most users of LC–MS instrumentation would not allow a bottle of triethylamine containing mobile phase to come near their instruments (there are exceptions to this, but we'll leave that topic for another column). So, how do we reconcile these differences in perspective, and when we consider adapting a method that involves triethylamine, how do we decide whether to leave it in or take it out?
The short answer is that advances in column technology have largely rendered triethylamine obsolete for analyses where it was once considered essential.
A longer answer requires a good understanding of how the characteristics of silicas used for LC stationary phases have evolved over the past couple of decades. In the 1990s, methods were developed for producing porous, spherical silicas for use as stationary phase substrates that have much higher purities compared to previous materials, particularly with respect to metal content (for example, iron and aluminum). This change in the quality of the silica was so pronounced that materials produced by the earlier methods are referred to as "type A," and materials produced using the more modern methods are referred to as "type B" (2). Perhaps the most consequential impact of the metal impurities present in type A silicas is that they significantly lower the acid dissociation constants of silanol groups on the surface of the silica particle. Figure 1 illustrates the loss of a proton (dissociation) from a surface Si-OH group yielding the negatively charged Si-O– group. With type A silicas starting at around pH 3, the fraction of silanols in the Si-O– form increases significantly (3). Below pH 3, the predominant form will be Si-OH, and above pH 3, the predominant form will be Si-O–. Whereas the Si-OH group can interact with analytes through hydrogen bonding interactions, the Si-O– group can interact with positively charged analytes through much stronger electrostatic (charge–charge) interactions. These strong electrostatic interactions can lead to poor peak shape, particularly when there is a relatively small population of these sites on the silica surface, and this usually leads to serious peak tailing (4). In the late 1970s, researchers discovered that adding alkylamines such as triethylamine to reversed-phase LC eluents at concentrations on the order of 50 mM dramatically improved peak shapes for amine-containing analytes (5). Over his years writing this "LC Troubleshooting" column, John Dolan often wrote about the uses of and problems with triethylamine (6). It is understood that, under conditions where the triethylamine is protonated (below about pH 10), the triethylammonium ion interacts strongly with the surface Si-O-– groups, effectively preventing analytes of interest from interacting with these sites and the expected poor peak shapes.


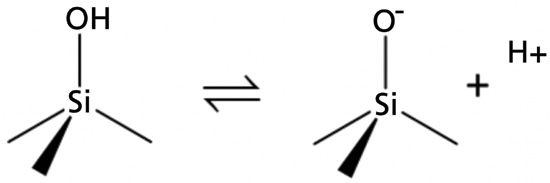
Figure 1: Illustration of isolated silica surface silanol dissociation.
The reduction in metal impurities in type B silicas results in an increase in the pH at which a significant fraction of silanols becomes deprotonated and increases from about 3 to about 7 (3). This means that most surface silanols on type B silicas will be protonated and neutral in mobile phases buffered at pH 6 or lower (for example, even in 0.1% formic acid in water). At this pH, the detrimental electrostatic analyte–silanol interactions described above are avoided. Thus, with many type B reversed-phase columns, excellent separations of amine-containing bases can be obtained with simple acidic mobile phases (for example, phosphoric acid, or ammonium formate adjusted to pH 4) that do not involve triethylamine.
When considering whether or not to continue using triethylamine, we should consider not only whether or not it is actually needed, but also ways that it may create more problems than it solves. For example, triethylamine can act as an ion-pairing reagent for anionic analytes, potentially increasing their retention in surprising ways (see below for more on ion-pairing). In another example, equilibration of reversed-phase columns in mobile phases without triethylamine after the column has been used with triethylamine can be very slow, and lead to significant selectivity changes over the course of days, or even weeks.
The bottom line is that if your environment requires continued use of a type A reversed-phase column, and the method was developed using triethylamine, then you are probably stuck with triethylamine for the life of the method. But, if you are adapting an older method that used triethylamine, but with a newer type B reversed-phase column, then it is worth taking the time to consider whether or not you really need triethylamine going forward. If you can get the separation you need with good resolution and peak shape, but without triethylamine, let it go!
Long-chain Ion Pairing Reagents: Take Them or Leave Them?
Historically, one of the weaknesses of reversed-phase separations has been low retention for hydrophilic compounds, particularly acidic and basic compounds for which retention drops precipitously when the compound is protonated or depronated, and becomes positively or negatively charged. For example, with a C18 column and a mobile phase containing 10% acetonitrile and 90% phosphate buffer at pH 2.4, the retention factor of benzoic acid is about 35. Under these conditions, the acid is protonated and neutral, and not very soluble in the mostly aqueous mobile phase. However, when the pH is raised to 6, benzoic acid deprotonates and becomes negatively charged, increasing the water solubility dramatically. In this case, the retention factor is around 1, a decrease of a factor of 35 compared to the condition where the acid is protonated and neutral (7). In the case of benzoic acid, there is still enough retention, even in the deprotonated state, to enable separation from other analytes, because the retention is influenced by the lipophilicity of the phenyl group. However, for other molecules where the rest of the molecule other than the ionogenic functional group is hydrophilic, it can be hard to get enough retention to develop a useful separation. Moreover, when analytes contain multiple cationic functional groups, interactions between these groups and the mobile phase may completely dominate retention, overwhelming any retention that might normally arise due to lipophilic parts of the molecule.
In the early days of liquid chromatography, this challenge of low retention for hydrophilic ionogenic compounds led to extensive use of what we refer to as ion-pairing reagents. These reagents are things like sulfonic acids (alkylsulfonates) and amines (alkylamines) with long (4 to 18 carbons) alkyl chains. The basic idea here is that the strong electrostatic interaction between the charged functional group of the ion-pairing reagent can form an ion pair with an analyte that has a functional group of the opposite charge. This ion pair has a lower water solubility than the unpaired analyte, which leads to higher retention under reversed-phase conditions. One can also find many examples of methods involving ion-pairing reagents as mobile-phases additives in vendor application notes, the peer-reviewed literature, and USP methods. For example, the USP method for pyridoxine (also known as vitamin B6, shown in Figure 2) calls for the use of hexanesulfonate in the mobile phase buffered at about pH 3 with acetic acid; here, the hexanesulfonate typically would be added to the solvent as a sodium salt. The pKa associated with the form of pyridoxine with the aromatic nitrogen protonated is about 5.6. This means that below pH 5.6, most molecules in solution will be positively charged, and above pH 5.6 (at least up to about pH 9), most molecules in solution will be neutral.

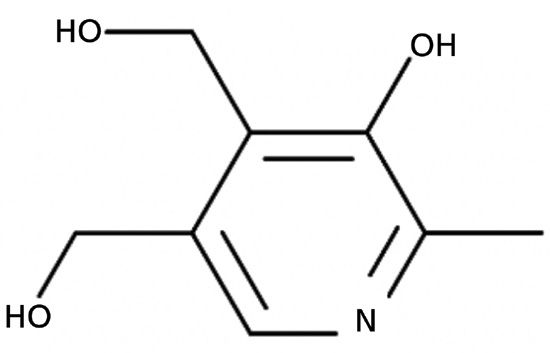
Figure 2: Structure of pyridoxine.
There is no question that these ion-pairing reagents provide an effective means of getting a "handle" on molecules that would otherwise be hard to retain on most reversed-phase phases without them. However, there are several reasons to avoid long chain alkylsulfonates and alkylamines in the modern chromatography laboratory. First, these ion-pairing reagents tend to be highly retained by reversed-phase phases themselves, which means that they do not instantly "go away" if we change from a mobile phase containing the ion-pairing reagent to one that does not (8). One way of thinking about this is that the ion-pairing reagent partitions into the stationary phase and stays there unless we take specific steps to wash it out. We refer to this as dynamic modification of the stationary phase, and, in the case of using an alkylsulfonate, the stationary phase will acquire some degree of negative charge when the ion-pairing reagent partitions into the stationary phase. In other words, the chemical characteristics of a reversed-phase column will be very different after it has been used with a long chain ion-pairing reagent, compared to when it was brand new. This kind of memory effect motivates many users to dedicate specific columns for use only with mobile phases containing long chain ion-pairing reagents. The second major reason to avoid these reagents in the modern LC laboratory is that they are not very compatible with mass spectrometry (MS), or at the very least are not MS-friendly. Alkylsulfonates are not volatile, and will quickly contaminate electrospray ionization sources. Long chain alkylamines are reasonably volatile, but are very difficult to remove from mass spectrometers after they have been introduced at the concentrations needed to have a useful effect on reversed-phase retention.
The good news here is that there are several viable alternatives to long chain ion-pairing reagents that can be used to increase retention of hydrophilic analytes. As with triethylamine as discussed above, if your environment does not allow you to change column chemistries or separation modes, then you are probably stuck with these ion-pairing reagents for the life of the method. However, if you are allowed some flexibility, then it is worth taking the time to consider whether or not continued use of long chain ion-pairing reagents really is warranted.
Alternative #1: Fluorinated Short Chain Ion-Pairing Reagents
Whereas hydrophilic acid mobile phase modifiers, such as phosphoric acid and formic acid, do not ion pair strongly enough with positively charged analytes to significantly increase retention on reversed-phase columns, fluorinated short chain carboxylic acids such as trifluoroacetic (TFA) and heptafluorobutyric acid (HFBA) can increase retention enough to be useful in this regard (9). Trifluoroacetic in particular does not cause any serious memory effects with reversed-phase columns, and is volatile enough to be used in with electrospray MS.
Alternative #2: Aqueous-Compatible Reversed-Phase Columns
Most major manufacturers of reversed-phase columns now sell specific chemistries that are advertised as "aqueous compatible" (10,11). Whereas with conventional C18 type stationary phases it is generally advised to avoid more than about 95% water in the mobile phase, these aqueous compatible phases (commonly referred to as AQ columns) can be used in 100% aqueous-mobile phases safely and effectively. The ability to go to a 100% aqueous mobile phase provides an avenue to increase retention of very hydrophilic molecules that would otherwise not be retained at all. For example, several small organic acids (such as acetic, tartaric, and succinic acids) can be readily separated using these phases (11). Finally, some phases designed for use with 100% aqueous mobile phases involve polar constituents (for example, polar endcapping ligands and polar embedded groups) that can interact with hydrophilic analytes through hydrogen bonding or electrostatic interactions.
Alternative #3: HILIC Separations
Although reversed-phase separations still dominate the LC separation landscape, users are becoming more comfortable with alternative separation modes, such as hydrophilic interaction (HILIC) separations, as the community continues to build up understanding about how and when HILIC works through fundamental research and new applications. HILIC and reversed-phase separations are complementary in many ways, not the least of which is retention for highly hydrophilic molecules. Although HILIC certainly will not replace all reversed-phase separations involving long chain ion-pairing reagents, it certainly is worth a try, particularly in situations where the long chain ion-pairing reagents are clearly undesirable for reasons such as those discussed above.
Tertiary Mobile Phase Mixtures: Take Them or Leave Them?
The final type of method we'll consider here is one that is focused on a very simple separation of one or two components, but involves a tertiary (or quaternary) mobile phase that specifies the use of two different organic solvents. Here again, numerous examples can be found in vendor application notes and USP methods. For example, the USP assay method for caffeine calls for a mobile phase containing 2.5% (v/v) acetonitrile and 2% (v/v) tetrahydrofuran (1). Now, it most definitely is the case that blending multiple organic solvents can be incredibly useful for adjusting selectivity in reversed-phase separations of multicomponent analyte mixtures (12). However, in assays that are focused on one or two constituents of the sample, the primary role of the organic solvent component of the mobile phase is retention control, which only requires one type of solvent. In the case of the caffeine method, the acetonitrile and tetrahydrofuran mixture could very easily be replaced with about 5% (v/v) acetonitrile, without affecting the assay results in any way. Moreover, this would simplify the method, make it more robust over the long term, and eliminate concerns around the use of tetrahydrofuran related to the potential for peroxide formation.
Summary
In this installment, we have discussed a few conditions commonly encountered when working with reversed-phase liquid chromatography methods that were established one or more decades ago, including the use of triethylamine and long alkyl chain sulfonates or amines as mobile-phase additives, and the use of organic solvent mixtures as mobile-phase modifiers. Although these approaches were certainly warranted in the past, advances in LC column technology have lessened the need for them. Understanding the origins of these approaches should help younger separation scientists in particular to decide which of these chromatographic legacies should be carried forward, and which of them can be let go when adapting methods developed in the past to meet current analytical objectives.
References
(1) United States Pharmacopoeia and National Formulary (USP 41-NF 36). (United States Pharmacopoeial Convention, Rockville, MD, 2016).
(2) L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography (John Wiley & Sons, Hoboken, New Jersey, 3rd ed., 2010), pp. 200–217.
(3) A. Méndez, E. Bosch, M. Rosés, and U.D. Neue, J. Chromatogr. A 986, 33–44 (2003). doi:10.1016/S0021-9673(02)01899-X.
(4) D.H. Marchand, L.R. Snyder, and J.W. Dolan, J. Chromatogr. A 1191, 2–20. (2008) doi:10.1016/j.chroma.2007.10.079.
(5) R. Gill, S.P. Alexander, and A.C. Moffat, J. Chromatogr. A 247, 39–45 (1982) . doi:10.1016/S0021-9673(00)84854-2.
(6) J.W. Dolan, LCGC North Am. 17, 100–106 (1999).
(7) D.R. Stoll, K. O'Neill, and D.C. Harmes, J. Chromatogr. A 1383, 25–34 (2015). doi:10.1016/j.chroma.2014.12.054.
(8) J.W. Dolan, LCGC North Am.14, 466–468 (1996).
(9) J. Dai, S.D. Mendonsa, M.T. Bowser, C.A. Lucy, and P.W. Carr, J. Chromatogr. A 1069, 225–234 (2005). doi:10.1016/j.chroma.2005.02.030.
(10) D.R. Stoll, LCGC North Am. 37(2), 80–90 (2019).
(11) D.R. Stoll, LCGC North Am. 37(2), 244–249 (2019).
(12) M.R. Euerby, F. Scannapieco, H.-J. Rieger, and I. Molnar, J. Chromatogr. A 1121, 219–227 (2006). doi:10.1016/j.chroma.2006.04.073.

Dwight R. Stoll

Dwight R. Stoll is the editor of "LC Troubleshooting." Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 50 peer-reviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC's editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com

Tony Taylor

Tony Taylor is the technical director of Crawford Scientific and ChromAcademy. He comes from a pharmaceutical background and has many years of research and development experience in small molecule analysis and bioanalysis using LC, GC, and hyphenated MS techniques. Taylor is actively involved in method development within the analytical services laboratory at Crawford Scientific and continues to research in LC–MS and GC–MS methods for structural characterization. As the technical director of the ChromAcademy, Taylor has spent the past 12 years as a trainer as well as developing online education materials in analytical chemistry techniques.













Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


