Data Integrity Focus, Part V: How Can USP Help Data Integrity?
LCGC North America
USP chapter can help ensure data integrity of a chromatography data system. Here’s how.
The updated version of United States Pharmacopoeia (USP) <1058> on analytical instrument qualification (AIQ) merges instrument qualification and computer validation into a single integrated process. We need to understand how the new version of USP <1058> can help ensure data integrity of a chromatography data system (CDS).
This is the fifth Data Integrity Focus article in a six-part series. The first presented and discussed a data integrity model to present the scope of data integrity and data governance program for an organization (1). The second part discussed data process mapping to identify data integrity gaps in a process involving a chromatography data system (CDS), and looked at ways to remediate them (2). The CDS was operated as a hybrid system, which was the subject of the third article (3), and the fourth part discussed how complete data and raw data mean the same thing for records created in a chromatography laboratory (4). In this fifth part, we look at the updated version of United States Pharmacopoeia (USP) <1058> on analytical instrument qualification (5), and see what the impact of this new general chapter has on data integrity.
Remember the data integrity model we discussed in Part 1 (1)? Immediately above the foundation layer was Level 1 that focuses on the right instrument or system for the job. This involves analytical instrument qualification (AIQ), either alone or in combination with computerized system validation (CSV). This is mirrored within USP <1058> and the data quality triangle where the base of the triangle is analytical instrument qualification. Thus, both the data integrity model and the USP <1058> data quality triangle (5) have AIQ as core to the integrity and quality of data. We will spend this part looking at how the new version of USP <1058> helps ensure the integrity of data generated, stored, and reported by a chromatography data system (CDS).
Impact of AIQ on Data Integrity
In the data integrity model, analytical instrument qualification and computerized system validation are essential to ensure the correct measurement by analytical instruments such as chromatographs and correct operation of software such as CDS applications. When you know that the instrument and software work as intended, you can have confidence that analytical procedures developed, validated, and applied in the layers above generate quality data where the integrity is ensured. However, before employing these procedures, let us look at some of the major regulatory citations involving chromatography data systems to see what we have to control and prevent data integrity violations.
A Parcel of Rogues
CDS have been at the heart of many data integrity violations since the Able Laboratories fraud case in 2005 (6). Table I lists the main regulatory citations abstracted from a number of US Food and Drug Administration (FDA) warning letters over the past 10 years against the three main areas of US GMP regulations (7). Note many of the instances of non-compliance listed in Table I can be adequately resolved by configuring the CDS application to protect electronic records, and to ensure that data storage can be restricted to specific folders in a database. For example, why would any user need deletion rights? Configuring all user types with no deletion rights in a CDS with an integrated database provides an additional benefit: you no longer need to search the system and audit trail for deletion events during a second person review.


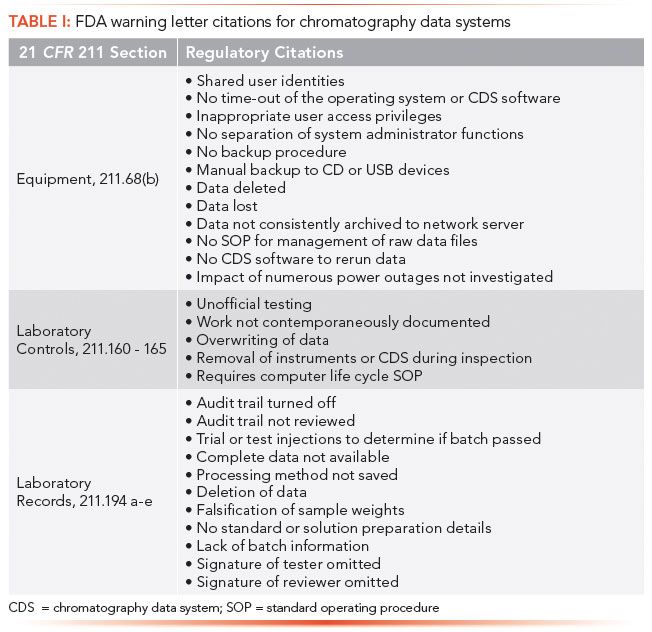
The table does not consider any other areas in a CDS, such as integration, that can affect data integrity. For background reading about the ideal CDS for a regulated environment including the need for a database instead of directories in the operating system, please see the four-part series on the topic published earlier in this magazine (8–11).
The USP <1058> Life Cycle Model
The starting point for considering how USP <1058> can help ensure data integrity is the life cycle model used by the past and current version of the chapter: the 4Qs model, which includes design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ). Readers familiar with the 2008 version of USP <1058> (12) may think that the 4Qs model is the same in both versions. Nothing can be further from the truth. There are significant changes that you need to be aware of that affect a laboratory's approach to AIQ. The first is the 4Qs life cycle model in USP <1058> (5), where there is only a linear textual description of each of the qualification phases. However, if presented as a V model, as shown in Figure 1, you start to see the relationships between each of the phases more clearly.

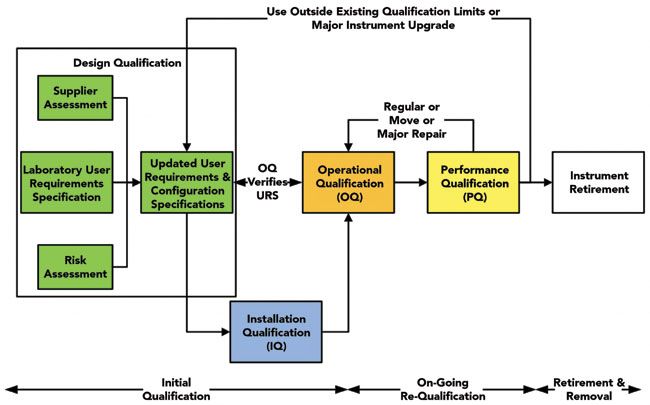
Figure 1: USP <1058> 4Qs life cycle model for integrated analytical Instrument qualification and computerized system validation (modified from [13]).
Look carefully at Figure 1. On the left-hand side, instead of a single box called design qualification (DQ), there is a separation of this phase into several parts. The first is for the laboratory to write a user requirements specification (URS) for the chromatograph and the CDS application software. The URS must be sufficiently detailed to define the intended purpose of the instrument and software, as required by GMP and GLP regulations (7,14). This URS is compared with the supplier's specification and software to confirm that the instrument and software are right for the job, and is typically updated to reflect the system being purchased. In addition, a system-level risk assessment should be conducted (15), as well as an assessment of the supplier that we will discuss later. When considering software, a URS is a living document and will need to be updated as you understand how the application functions and also to document the configuration settings of the application, as shown in Figure 1.
This portion of the updated 4Qs model is harmonized with clauses 3.2 and 3.3 of European Union Good Manufacturing Practices (EU GMP)Annex 15 on Validation and Qualification (16) which states that there needs to be a URS (clause 3.2) and that the URS must be compared with the selected system (clause 3.3). Further harmonization with Annex 15 clause 2.5 (16) is the ability to merge qualification documents, such as installation qualification (IQ) and operational qualification (OQ) protocols, where practicable. This has the advantage that only a single IQ/OQ protocol needs pre-execution review and post-execution approvals by the laboratory and quality assurance (QA) staff. The main changes in the updated version of USP <1058> can be found in a "Questions of Quality" column in LCGC Europe (17), as well as in a three-part detailed discussion of the impact of the new general chapter on data integrity of chromatographic instruments by Paul Smith and me (18–20).
USP <1058> and Software Validation
What does USP <1058> say about software validation? Compared with the old version (12), a lot more. The first point is:
There is an increasing inability to separate the hardware and software parts of modern analytical instruments. In many instances, the software is needed to qualify the instrument, and the instrument operation is essential when validating the software. Therefore, to avoid overlapping and potential duplication, software validation and instrument qualification can be integrated into a single activity (5).
Put simply, do one job of validating the whole system, rather than two separate tasks that consist of qualifying the instrument with the unvalidated CDS software, and then validating the application with the qualified instrument.
USP <1058> Software Risk Assessment
When considering Group C (instruments with application software for data acquisition, interpretation, and reporting), there is a risk assessment as shown in Figure 2, where there are three types of software discussed. As shown in Figure 2, these three types can be easily mapped to the Good Automated Manufacturing Practice (GAMP) software categories (21) as either category 3 (type 1), category 4 (type 2), or category 4 with category 5 modules (type 3). CDS software is category 4 (22) or USP <1058> Group C type 2.

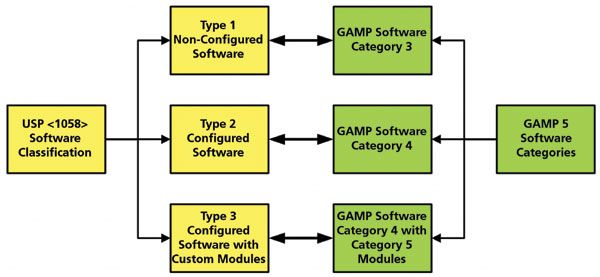
Figure 2: USP <1058> Group C software types mapped to Good Automated Manufacturing Practice (GAMP) software categories.
Although the overall CDS application is category 4 (as the business process can be changed, for example), implementing electronic signatures, many of the functions within the software are category 3, for example instrument controls that are only parameterized (22).
Leveraging the Supplier's Work
USP <1058> (5) and GAMP 5 (21) both suggest further risk assessment:
The user can apply risk assessment methodologies and can leverage the supplier's software testing to focus the OQ testing effort (5).
If an organization can find out what a CDS supplier has done during the development and testing of a version of the software, this can be used to justify reducing the amount of testing required in the laboratory validation. The general chapter makes the following statement:
The supplier of the system should develop and test the software according to a defined life cycle and provide users with a summary of the tests that were carried out. Ideally, this software development should be carried out under a quality management system (5).
To help reduce the amount of work, you should carry out a supplier assessment, either on site or remote, to ensure that all category 3 functions in the CDS software have been adequately specified and tested, so that you can state that they are fit for intended use. A postal questionnaire is not the vehicle for this assessment. Linked with the assessment should be a list of functional tests carried out on the released software by the supplier to further justify your overall validation approach. In this way, much basic validation work can be reduced, and you can focus on the real validation stuff, as we shall now see.
CDS Validation Approach
On software validation, USP <1058> further states that:
Where applicable, OQ testing should include critical elements of the configured application software to show that the whole system works as intended. Functions to test would be those applicable to data capture, analysis of data, and reporting results under actual conditions of use as well as security, access control, and audit trail.
...When applicable, test secure data handling, such as storage, backup, audit trails, and archiving at the user's site, according to written procedures (5 [emphasis added]).
Note the two words in bold in the quotation above: configured application. OQ testing of the whole system should be conducted with the application software configured. What does that mean in practice? CDS application software used in regulated laboratories has a number of functions that can be turned on or off or set (configured), and these must be documented and are part of the definition of intended use, along with the user requirements specification, as seen in Figure 1.
Configuring the CDS Application to Ensure Data Integrity
To overcome most of the regulatory issues cited in Table I, the CDS software needs to be configured to protect electronic records, enforce working practices, segregate duties including avoiding conflicts of interest, and work electronically with electronic signatures. These software settings or policies must form part of the intended use of the system, and must be documented either in a configuration specification or the URS, as shown in Figure 1.
Note that other areas of system configuration, such as custom calculations and reports, are not included in this configuration specification, as these can be appropriately controlled with SOPs (22). This approach provides more flexibility, because it avoids the need for constantly updating the more extensive URS or configuration specification documents. Make no mistake, CDS custom reports and calculations must be appropriately specified, built, and tested, but they can be controlled via procedures in a much simpler way compared with a system validation.
Supplier OQ and User OQ Testing?
One of the constraints with the USP <1058> 4Qs model is the lack of granularity and one of the impacts is with the OQ phase. Typically, a supplier OQ is conducted on the unconfigured software to demonstrate that the software and the chromatographs interfaced with it work correctly. This supplier OQ is an essential part of supplier installation and checkout work before handover to the laboratory. However, as shown above, USP <1058> requires that the configured CDS application be tested against the URS in the OQ (5).
Therefore, a second phase of OQ testing against the URS and configuration specification is conducted. This testing in computerized system validation (CSV) is known as user acceptance testing (UAT), and this demonstrates fitness for purpose of the overall system. It is essential that tests be conducted to demonstrate:
- security and access control for the various user types
- intended use chromatographic analysis, processing, and reporting with electronic signatures
- calculations, for example, calibration models, and system suitability tests (SSTs) actually used by the laboratory are correct
- adequate size or capacity of the system
- data integrity functions such as protection of electronic records and audit trail work as configured.
- More detail of this can be found in a book on validation of chromatography data systems (22).
Summary
We have focused on the requirements of the updated USP <1058> on analytical instrument qualification and how it can help with ensuring the integrity of data generated by a chromatography data system. In the sixth and last part of this article series on data integrity, we will look at the people who will be policing and providing oversight of the whole data integrity process. Enter stage left, those intrepid people from quality assurance.
References
(1) R.D. McDowall, LCGC North Am. 37(1), 44–51 (2019).
(2) R.D. McDowall, LCGC North Am. 37(2), 118–123 (2019).
(3) R.D. McDowall, LCGC North Am. 37(3),180–184 (2019).
(4) R.D. McDowall, LCGC North Am. 37(4), 265–268 (2019).
(5) USP <41> General Chapter <1058> Analytical Instrument Qualification (United States Pharmacopoeia Convention Rockville, MD, 2018).
(6) Able Laboratories Form 483 Observations, 2005, 1 Jan 2016, URL: http://www.fda.gov/downloads/aboutfda/centersoffices/officeofglobalregulatoryoperationsandpolicy/ora/oraelectronicreadingroom/ucm061818.pdf.
(7) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration: Sliver Springs, MD, 2008).
(8) R.D. McDowall and C. Burgess, LCGC North Am. 33(8), 554–557 (2015).
(9) R.D. McDowall and C. Burgess, LCGC North Am. 33(10), 782–785 (2015).
(10) R.D. McDowall and C. Burgess, LCGC North Am. 33(12), 914–917 (2015).
(11) R.D. McDowall and C. Burgess, LCGC North Am. 34(2), 144–149 (2016).
(12) USP <31> General Chapter <1058> Analytical Instrument Qualification (United States Pharmacopoeial Convention: Rockville, MD, 2008).
(13) P. Smith and R.D. McDowall, LCGC Europe 28(2), 110–117 (2015).
(14) 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies (Food and Drug Administration: Washington, DC, 1978).
(15) C.Burgess and R.D. McDowall, Spectroscopy 28(11), 21–26 (2013).
(16) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation (European Commission, Brussels, 2015).
(17) R.D. McDowall, LCGC Europe31(1), 36–41 (2018).
(18) P. Smith and R.D. McDowall, LCGC Europe 31(7), 385–389 (2018).
(19) P. Smith and R.D. McDowall, LCGC Europe 31(9), 504–511 (2018).
(20) P. Smith and R.D. McDowall, LCGC Europe 32(1), 28–32 (2019).
(21) Good Automated Manufacturing Practice (GAMP) Guide Version 5 (International Society for Pharmaceutical Engineering, Tampa, FL, 2008).
(22) R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, United Kingdom, 2nd Edition, 2017).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com















Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



