Method Reproducibility — A Case Study
LCGC Europe
Excess variability is not acceptable in a pharmaceutical method.
This month's instalment of "LC Troubleshooting" examines an issue associated with a liquid chromatography (LC) method for a pharmaceutical product. Because the method is proprietary, some of the details will be left out of the method description presented here. This method had performance requirements that were typical for such methods — the average assay value of several replicate injections had to fall within 98–102% of the expected value. The method range was 70–130% of the nominal value for the drug in the product. The product contained two active ingredients, which will be referred to as compound A and compound B for the present discussion. The issues encountered with this method provide a good example for the application of general troubleshooting principles.
The Method
The method used a 150 mm × 4.6 mm reversed-phase column packed with 5 µm diameter particles and maintained at 30 B0C with a column oven. The mobile phase was 90:10 phosphate buffer–acetonitrile with a flow-rate of 2 mL/min. UV detection was used.
The method had been validated several years ago and was transferred to a second laboratory recently. When adjustments were made to the method for a new application, the performance requirements for compound B were easily met. Whereas, compound A passed at concentrations of 100% of the nominal value and below, but it did not meet the ±5% specification window at 130% concentration (95%). It was found that reducing the injection volume from 20 µL to 10 µL allowed the 130% concentration to meet the requirements for both compounds, so the method was revalidated for 10 µL injections. Typical results at the 130% level are summarized in Table 1.
Are the Results Reasonable?
One of the first steps to take when examining any problem related to the results of an LC method is to determine if the data obtained are reasonable for the method conditions. Three parameters that are examined easily are retention time, peak width, and peak shape. Let's look at each of these in turn.
Retention: Rather than look at retention times alone, it usually is more informative if we check the retention factor, k. For the best chromatographic performance, it generally is desirable to have k-values between 2 and 10, although most workers will settle for values of 1–20 if necessary. The retention factor is calculated as


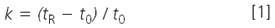
where tR and t0 are the retention time and the column dead time, respectively. The column dead time can be measured by injection of an unretained compound, such as uracil, but for many purposes, an estimate is good enough. An estimate of t0 for 4.6 mm i.d. columns is


where L is the column length in millimetres and F is the flow-rate in millilitres per minute. So for our 150 mm × 4.6 mm column, t0 ≈ (0.01)(150)/2 = 0.75 min. This now allows us to calculate k-values for the results of Table 1 as k = 2.2–3.1 (compound A) and 4.1–5.8 (compound B), all of which are within acceptable limits. Reproducibility can be more of a problem when k < 1 is observed, but it looks like solute retention is reasonable for both peaks under all three conditions.
Peak width: Although you can compare absolute peak widths between runs, it is hard to evaluate if the widths are reasonable or not. A better approach for isocratic separations is to calculate the column plate number N instead. This should be relatively constant for all peaks in the chromatogram. The plate number can be calculated as

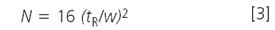
where w is the peak width measured at the baseline between lines drawn tangent to the peak. (The plate number also can be determined in the same manner by using the peak width at half the peak height and multiplying by 5.54 instead of 16). What is a reasonable value of N? For a 5 µm particle column, manufacturers' test chromatograms often report 80000–100000 plates/m, but these measurements are made under ideal conditions with well-behaved test compounds. A good estimate for real compounds under typical operating conditions is


where dp is the particle diameter in micrometres. So for our 150 mm, 5 µm column, we expect N ≈ 9000; if we observe values within approximately 20% of this, the column is giving acceptable performance for routine methods. The data of Table 1 suggest that the plate number was a bit low on the original system, but the new one gives N > 7000 in all instances, so this is acceptable.
Peak tailing: Ideally, peaks should be perfectly symmetrical, but from a practical standpoint, they never are. Peak tailing is measured as the USP tailing factor:

where wtotal is the total peak width and wfront is the width of the front half of the peak (front edge to perpendicular dropped from the peak apex), measured at 5% of the peak height. If TF < 1.5, one seldom is concerned about peak tailing. All of the tailing factors listed Table 1 are ≤ 1.2, so peak tailing is not an issue with this method.


Table 1: Typical results at 130% of the nominal concentration.
Compare the Results
Now that we've examined the basic chromatography parameters and found them satisfactory, we can dismiss them as a likely source of the problem. Next, we should compare the results of the different run conditions. We are not too concerned with the shift in retention — this is attributed to a change in flow-rate and column temperature between the two methods. However, the large change in response (almost a 30-fold increase) is a potential area of concern. The >50% increase in plate number between the original method and the current implementation could be important or might just reflect a change in equipment, plumbing, or column technology. Let's take a closer look at the detector response.
Detector response: Although many of today's UV detectors boast linearity of >2 absorbance units (AU) in their specification sheets, many workers are much more comfortable when the signal is <1 AU. As the peak gets large enough to saturate the detector, the top tends to flatten out, giving nonlinear behaviour. So the when the current method using a 20 µL injection gives a peak for Compound A that is 1.9 AU (1900 mAU) tall, it is worth checking to be sure the detector linearity is not being exceeded. To check this, one can reduce the amount of sample on column by reducing the sample concentration, the injection volume, or both. The reduction of the injection volume is the simplest approach, and it was tried here. With a 10 µL injection, the peak height is now 0.9 AU, less than the preferred 1 AU limit. The average assay value now passes specifications, so this suggests that the problem was related to too large a signal. Note that further support for this conclusion comes from looking at the data for compound B. In both the 20 µL and 10 µL runs, the average assay value passes the specifications and the peak is <1 AU tall in both instances. Finally, the conditions for the original method were such that the peak height was only 70 mAU, so this was well below the region of potential detector overload, and acceptable performance was observed. Before we draw a final conclusion about the detector response as the problem source, let's take a closer look at the retention and plate number data.
Overload: Injection overload has to do with injecting a volume of sample that is too large or using an injection solvent that is too strong, or both. The sample was dissolved in mobile phase in the present situation, so the injection solvent strength should not be an issue. A useful rule of thumb for the allowable injection volume of sample dissolved in mobile phase is to keep the injection volume ≤15% of the peak volume. We can figure out the allowable volume from the data at hand. We can solve Equation 3 for the peak width (in minutes) and convert this to volume by multiplying by the flow-rate (2 mL/min). The width of the peak for compound A in the 10 µL run is about 290 µL, which means that we should be able to safely inject as much as 45 µL without concern of injections that are too large. Thus, it is unlikely that the 20 µL injection is too large. We can dismiss injection volume or injection solvent strength as likely candidates for our problem.
Mass overload is related to the mass of sample that can be injected before the stationary phase is sufficiently loaded to cause changes in the chromatography. A rule of thumb for this is that most columns will handle about 10 µg of sample per gram of packing material before the column is overloaded (but this does vary with the sample type and column). This translates to 10–20 µg of sample on column for a 150 mm × 4.6 mm column. In the present instance, the sample concentration was 1 mg/mL, so a 20 µL injection would be 20 µg on column. This is close to the expected column capacity, therefore, the reduction of the injection volume, so that 10 µg was injected instead of 20 µg, could make the difference between overload or not. Three symptoms of column overload are the reduction of retention time, an increase in peak width and more pronounced peak tailing. None of these symptoms show up when the results for compound A or B are compared between the 10 mL and 20 mL injections, so column overload is unlikely.
Conclusions
The data we examined pointed to a possible detector overload. What could have changed between the original conditions and the ones currently used? The original system might have had excessive extracolumn volume in the form of large-diameter tubing, different fittings, or a large volume detector cell. These factors might have contributed to the lower plate number (and, thus, broader peak) and smaller peak height.
With some LC troubleshooting problems, we may not have the luxury to fully identify the root cause of the problem. A suitable solution was found for the present example, and a likely cause was identified. The orginal method continued to work acceptably for its intended application and the modified (and revalidated) method also worked as intended. This is a good example of the importance of carefully examining method performance to determine if an existing method will work for a new application.
Nastaran Sigari is a senior research scientist at a pharmaceutical company. She has spent 21 years working for several major pharma companies, working on development and validation of stability indicating assays as well as troubleshooting LC methods. "LC Troubleshooting" editor John W. Dolan is vice-president of LC Resources, Walnut Creek, California, USA; and a member of the Editorial Advisory Board of LCGC Europe. Direct correspondence about this column to "LC Troubleshooting", LCGC Europe, Advanstar House, Park West, Sealand Road, Chester CH1 4RN, UK.
Readers can also direct questions to the Chromatography Forum at http://www.chromforum.com
Erratum
In December's LC Troubleshooting (LCGC Eur., 19(12), 649), Table 1 contains a typographical error. The units on the right-hand column should be µL/cm not mL/cm.















University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.





