A LEAN Approach for the Determination of Residual Solvents Using Headspace Gas Chromatography with Relative Response Factors
Special Issues
This study demonstrates a new LEAN approach and method, where 25 solvents can be simultaneously determined based on predetermined relative response factors (RRFs) against an internal standard (decane) with only one injection of sample solution.
This study demonstrates a new LEAN approach and method, where 25 solvents can be simultaneously determined based on predetermined relative response factors (RRFs) against an internal standard (decane) with only one injection of sample solution. The RRF average value of each solvent was determined by comparison of slope against internal standard from linearity experiments in the range of 10–200% and the response factor against internal standard at the ICH Q3C limit based on nominal sample concentration. Validation was performed successfully covering specificity, linearity, sensitivity, precision, accuracy, stability, and robustness. Laboratory efficiency and instrument utilization rates were significantly improved, leading to time and cost savings of more than 60% compared to a conventional external standard headspace gas chromatography (HSâGC) method since this method was implemented in 2014. It has been demonstrated that this generic RRFâbased HS-GC method is a LEAN approach for residual solvents testing in pharmaceutical substances, from process controls to quality control (QC) analysis.
Organic solvents are routinely used in the synthesis and manufacture of drug substances, excipients, and drug products. They can also be formed as by-products or side reactions during the manufacturing process. Their use or presence is therefore an inherent part of the pharmaceutical manufacturing process. However, as residual solvents do not provide any therapeutic benefits, can potentially impact the physicochemical properties of substances, and can even pose a potential safety risk to the patient, they should be controlled as much as possible to meet ingredient and product specifications, good manufacturing practices, or other quality-based requirements (for example, quality guidelines ICH Q3C (R6) [1] and USP <467> Residual Solvents [2]), which regulate the solvent’s classification and control limits based on the toxicity.
Specific methods (nuclear magnetic resonance [NMR], gas chromatography [GC]) and nonspecific methods (loss on drying [LOD] and thermal gravimetric analysis [TGA]) are often used to determine the solvent residues based on the intended use in the pharmaceutical industry. Among all the methods, headspace (HS)-GC–flame ionization detection (FID) has been the most popular instrumental setup in the pharmaceutical industry because of its high sensitivity and low matrix effects. Generic and fast methods were developed and published from various suppliers and laboratories, which readily facilitated the determination of residual solvents in pharmaceutical substances (2–8).
In the traditional approach, residual solvents are determined by external standard methods using reference standard(s) containing the solvents of interest. This approach is selective and accurate; however, as the value stream map of traditional residual solvents determination using external standards shows (Figure 1), it requires frequent sample and standard preparation. For example, when considering one sample with three interested solvents, the net working time is about 90 min (run and data analysis excluded). Therefore, it is not the preferred solution for the early phase R&D laboratory because of the dynamic needs and short time demands.


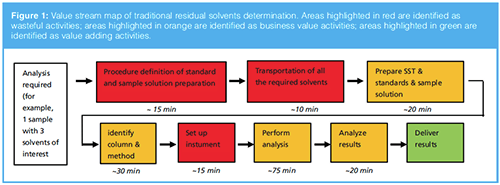
Scientists from AstraZeneca R&D developed an intelligent GC–FID (direct injection) relative response factor (RRF) method where predetermined RRFs were used to quantify the solvents of interest (3). It significantly improved the laboratory efficiency; however, the application was not further disclosed if it was routinely used in a regulated environment (for example, GMP) and there is the potential risk of carryover and matrix effects for sample analysis, and also lower column robustness and fouling because direct injection was used for this method.
Headspace GC is considered a more suitable technology for the determination of residual solvents in nonvolatile materials, such as drug substances and drug products because only volatile components are introduced into the GC system, potential contamination to the system can be reduced, and therefore robustness can be enhanced (4).
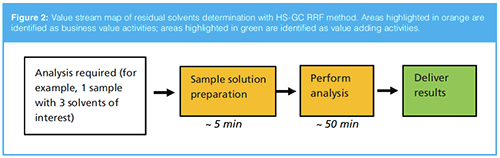
A new generic HS-GC RRF method was developed and further validated under GMP in our R&D laboratories. The process flow is shown in Figure 2. One sample with three interested solvents was again considered; the net working time is only about 5 min (run time and data analysis excluded), which meant that more than 75% of the sample and standard preparation time was saved compared with the traditional method. The study results demonstrated that the method is a fast and economical approach for residual solvents testing from in-process monitoring and controls to end product quality checks. The method is designed to be suitable for the determination of residues in a range of common processing solvents, including newer processing solvents used in “green chemistry” initiatives.

Experimental
Reagents and Chemicals: The solvents were analytical grade or above and purchased from commercial resources.
Instrumentation: An Agilent 7890A GC equipped with a flame ionization detector and a G1888 Head Space sampler was used. GC-HS system control, data acquisition, and processing were all accomplished by Chromeleon software (Thermo Fisher Scientific). The GC column employed was a 30 m × 0.32 mm,1.8âµm Agilent J&W DB-624 (6% cyanopropylphenyl and 94% dimethyl polysiloxane) fused silica capillary column. Headspace sample vials of 20-mL size were utilized with a Teflon-lined septum and an aluminium crimp cap (Agilent).
Final Method Operating Conditions:
GC Conditions:
Inlet temperature: 200 oC; injector mode: split; split ratio: 20:1; carrier gas: helium (or hydrogen). If H2 is used as carrier gas, the retention times are slightly shifted forwards in comparison to He; flow: 2.0 mL/min; mode: constant flow; vial pressurize: 14.0 psi; oven temperature program: 50 oC hold for 3 min, ramped to 80 oC with 5 oC/min, and finalize to 230 oC with rate 30 oC/min, hold for 2 min; detector: FID; detector temperature: 300 ºC; H2 flow: 40 mL/min; air flow: 400 mL/min; make up flow: 30 mL/min.
Headspace Parameters:
Oven temperature: 120 oC; loop temperature: 130 oC; transfer line temperature: 135 oC; vial equilibration time: 10 min; mixing speed: low; pressurization time: 0.5 min; loop fill time: 0.25 min; loop equilibration time: 0.1 min; injection time: 1.0 min; cycle time: 23 min; headspace vial: 20 mL.
Solution Preparations: Solution for GC analysis, 1 mL of solution in 20 mL headspace vial.
Internal Standard Solution:
The internal standard solution (decane in N-Methyl-2-pyrrolidone [NMP]) was accurately prepared at about 0.05 mg/mL by diluting decane with NMP.
Sample Solutions:
Approximately 50 mg of sample was dissolved by adding 1 mL of internal standard solution into a 20-mL headspace vial, sealed with the cap and ready for injection.
Reference Solution ( = Specificity Solution):
Each solvent was accurately prepared at the level equivalent to the appropriate ICH Q3C limit by dilution with internal standard solution based on nominal sample concentration 50 mg/mL. Taking ethanol as an example, where the ICH Q3C limit is 5000 ppm, the appropriate concentration for ethanol to be prepared is 0.25 mg/mL ( ï½ 5000 ppm*50 mg/mL/106). For methylisobutyl ketone (MIBK), the draft limit of 2250 ppm from the ICH residual solvents Q3C(R6) guideline, step 2 (for consultation and discussion) was used to define the limit. The finalized revision (ICH Q3C(R6) Current Step 4 version dated 20 October 2016) moved MIBK from Class III to Class II solvent with a limit of 4500 ppm (PDE: 45 mg/day) (1).
Reporting Limit (RL) Solution ( = 10% of Reference Solution):
The reporting limit solution was prepared by 10-fold dilution of the reference solution with internal standard solution.
System Suitability Test (SST) Solution:
A mixed SST solution containing reporting level of methanol, 2-butanone, ethyl acetate, toluene, decane, and 1, 2-dimethoxyethane at 20% of reference solution was prepared.
Determination of Relative Response Factor (RRF) and Calculation:
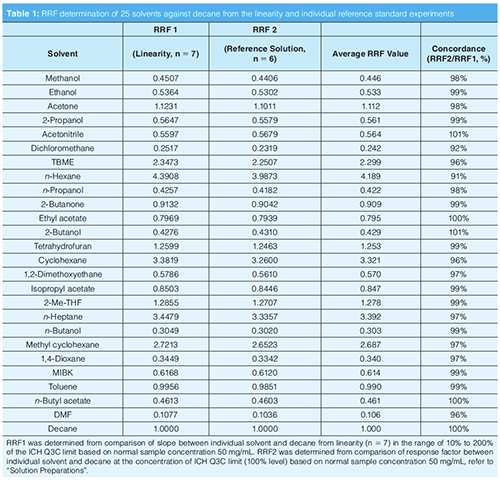
A mixture of 25 reference solvents and decane was prepared for RRF determination in NMP. The concentration of each solvent was calculated according to the individual concentration limit in ICH Q3C (step 2) based on a nominal sample concentration of 50 mg/mL. There were two approaches used for the RRF determination and average value was used for calculation. First, RRF1 determination, the RRF of each solvent to decane (internal standard) was determined by comparison of slope from linearity experiments, with solvents in the range of 10% to 200% of the ICH Q3C limit (for example, 0.025 mg/mL to 0.5 mg/mL for ethanol) and decane at 0.01% to 0.2% (m/m) (equation 1, RRF1). Second, RRF2 determination, the relative response factor was determined by comparison of response factor of each solvent to decane from six injections of reference solution at a single concentration (equations 2 and 3), average RRF (equation 4) was calculated for residual solvents determination (equation 6).
RRF1 = Ssolvent/SIS [1]
RFsolvent = Asolvent/Csolvent [2]
RRF2 = RFsolvent/RFIS [3]
Where, Ssolvent = slope of regression equation from validation for each solvent; SIS = slope of regression equation from validation for decane; Asolvent = average peak area of solvent from GC chromatogram (n = 6), pA; Csolvent = concentration of solvent, mg/mL; RFsolvent = response factor of solvent, pA/(mg/mL); RFIS = response factor of internal standard (decane), pA/(mg/mL).
RRF = (RRF1+RRF2)/2 [4]
Concordance (RRF agreement): = RRF2/RRF1*100% [5]
Csolvent (ppm) = (As × Cdecane)/(Adecane × Cs × RRF) ×106 [6]
Where, Csolvent = amount of solvent in sample, ppm; As = peak area of solvent in sample solution, pA; Cs = concentration of sample solution, mg/mL; Adecane = peak area in sample solution, pA; Cdecane = concentration of decane in solvent, mg/mL; Cs = sample concentration, mg/mL.
Results and Discussion
Method Development and Proof of Concept: The idea was to develop a fast method to address business needs focused on speed and efficiency. RRF-based approaches fit well for this purpose: 1) predetermine the relative response factor of each solvent to an internal standard; 2) dissolve sample in internal standard solution; 3) perform GC analysis (≥ 1 injection of sample) and results calculation based on RRF against the internal standard. With this concept, the method was developed.
Internal Standard Selection:
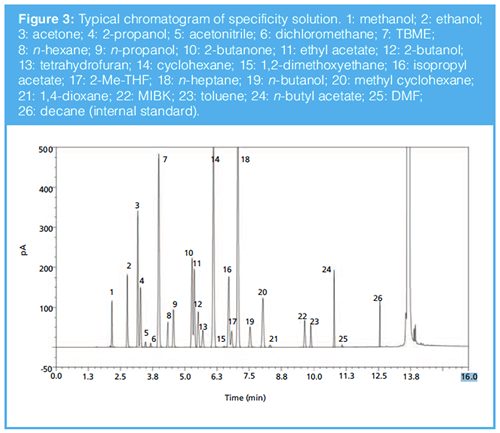
In principle, a good internal standard for HS-GC measurement should have the following properties: good response and peak shape, no significant carryover, stable and chemically inert, pure, relatively inexpensive, commercially available, minimal chance to overlap with the target analytes, and should also not be potentially present in the samples of interest. Decane and toluene were assessed during method development and finally toluene was ruled out because it was often used in process development and manufacturing processes and could be potentially present in the samples of interest. Decane was selected as it meets all these criteria and elutes later in the chromatogram (Figure 3). It is well separated from all the solvents of interest, is not commonly used as a processing or manufacturing solvent, and perfectly fits as an internal standard for the purpose of residual solvents determination.

As decane is a high boiling solvent, the potential carryover was examined by repeated injections of the decane internal standard solution (0.05 mg/mL decane in NMP) and blank solvent (NMP). The results show that no significant residue was found in the blank solution following the repeated injection of internal standard solutions and this confirmed decane as a good option as internal standard. Furthermore, this low carryover was later demonstrated and confirmed during routine sample testing.
Diluent and Solvents Selection:
A good diluent (sample solvent) for residual solvents determination is expected to have the following properties: good solubility for the samples, pure, well separated with the solvents of interest, stable and chemically inert and not susceptible to degradation under the operating conditions, relatively inexpensive, and readily commercially available. Dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and NMP were investigated as potential diluents. It was found that all three diluents could provide similar solubility, but NMP could accommodate more solvents for analysis because it elutes after DMF and DMSO in the developed GC conditions. In addition, given the fact that impurities in DMF and DMSO were interfering with decane (internal standard), NMP exhibited a “cleaner” chromatogram and was finally selected as the diluent. The most commonly used solvents in process development were identified prior to developing the analytical method. In total, 25 solvents (classes 2, 3, and some unclassified solvents) were in scope. Since class 1 solvents are highly carcinogenic or toxic, they are generally avoided in pharmaceutical manufacturing and are therefore not in the scope of the method requirements.
Headspace and GC Operating Conditions: Selection and Development:
GC method operating conditions were developed based on the wellâestablished and available internal generic method and key parameters, for example, the oven temperature gradient was adjusted by evaluating the resolution of specificity solution, sensitivity of RL solution, and total analysis time. As shown in Figure 3, all the solvents were well separated within 16 min, the final optimized conditions are shown above.
The headspace conditions were developed by considering the following points: ensure elution of DMF, which is the highest boiling solvent of interest; have a good response for all the solvents of interest; and avoid potential carryover during analysis. The finalized conditions are shown in the experimental section. The selected HS-GC operating parameters are within the typical GC parameters for a generic separation of residual solvents reported in previous publications (5).
Determination of RRF Value and Proof of Concept:
After the method operating conditions were established, the RRF was determined by two approaches (for calculation formula refer to Determination of Relative Response Factor [RRF] and Calculation) and the average value was used as a working RRF in the final method. The concordance between the RRF values determined by the two approaches was acceptable for all solvents of interest. The concordance is generally higher for class 3 solvents where precision is better owing to the higher concentration ranges used for the linearity and reference solution. Conversely, the RRF concordance is generally lower when the precision is lower, for example, for non-class 3 solvents where concentration levels and reporting levels are lower and solvents such as DMF, DCM show the lowest detector response. This relationship of RRF concordance to precision is also supported by the results shown in Table 1.

Since the RRF is the key to method accuracy, the RRF value was investigated in a different laboratory on a second instrument (same brand and model) during method development, validation, and transfer, and consistency between each analysis was shown.
With the established method and RRF value, several laboratory samples with known content of solvent residue calculated by external standard methods were also analyzed and consistent results were obtained. Therefore the concept and suitability of the RRF-based method was proven.
System Suitability Test:
To ensure the delivery of accurate, reliable, and consistent results, the suitability of the HS-GC system should be checked periodically, or even in each analysis sequence, for example, for a GMP analysis. Considering the ultimate goal of GMP application, the worst-case scenario was taken to assess the system suitability. A mixture of solvents including methanol, 2-butanone, ethyl acetate, toluene, and decane at reporting limit levels and 1, 2-dimethoxyethane at double the reporting limit (named SST solution) was proposed to check the system performance. The selected solvents were located at early, middle, and late positions (time windows) in the chromatogram and are good indicators to confirm the method specificity and retention times. 2-butanone and ethyl acetate represent the worst separation and most critical resolution (resolution 1.1, criteria >1.0) and 1, 2-dimethoxyethane (signal-to-noise ratio [S/N] 15, criteria >10) shows the lowest response and is an effective measure of sensitivity.
To confirm the absence of any potential interference from the sample, the variation between the decane peak area in the internal standard in blank solution (NMP) and sample solution was also proposed as a system suitability test. The acceptance criteria was proposed as variation < 10%, considering the variability from sample preparation and instrument capability.
Method Validation: The method was validated according to the ICH Q2 (R1) guideline by evaluating the specificity, reporting limit (sensitivity), precision, linearity, accuracy, stability, and robustness.
Specificity:
The specificity of this method was evaluated by injecting a solution containing all the solvents of interest at the ICH Q3C limit based on nominal sample concentration 50 mg/mL (named reference solution). As shown in Figure 3, all the solvents of interest were sufficiently separated from each other, and their identity was confirmed by injection of individual solutions. The developed method was found to be sufficiently selective.
Reporting Limit and Limit of Quantification (LOQ):
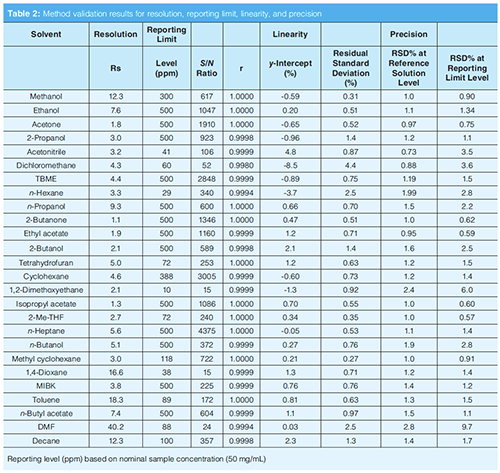
Limit of quantification (LOQ) is defined as the lowest amount of analyte in the sample that can be quantitatively determined. The reporting limit is equal or greater than LOQ and was confirmed by the S/N of each solvent in RL solution (10% concentration of reference solution). The results are shown in Table 2, and all S/N ratios of each solvent at the RL level are ≥ 10. The reporting limit of all solvents was defined at 10% of the individual concentration limit based on a nominal sample concentration 50 mg/mL.

Linearity:
The linearity of peak response versus concentration was studied from 10% to 200% of reference solution at seven concentration levels (10%, 30%, 60%, 80%, 100%, 120% ,and 200%). The linearity of decane (internal standard) was studied from 0.01% to 0.2% (m/m). A simple linear regression analysis by the least squares was applied for each solvent. The value of correlation coefficient (r), y-intercept (in % relative to the calculated response at reference solution), and residual standard deviation (RSD, relative to the concentration of each solvent in reference solution) were evaluated and criteria were set as r ≥ 0.98, y-intercept ≤25%, and RSD ≤ 10%, respectively. As shown in Table 2, all the linearity parameters meet the requirements and good linearity for each solvent was demonstrated. Slope (a) of the regression line was used for RRF1 calculation.
Precision:
Method precision was assessed by the RSD of the peak area on six injections of reference solution (at relevant ICHQ3C limits) and reporting limit solution (approximately 10% of relevant ICHQ3C limits). Six replicate measurements were performed and RSD% of the area was calculated. For reference solution and RL solution, RSD of the peak area for each solvent was in the range of 0.7–2.8% for the reference solution and 0.6–9.7% at the reporting limit solution (Table 2). The precision is the lowest for non-class 3 solvents, where levels and reporting levels are lower, in particular for those that show the lowest detector response (for example, DMF, 1,2-Dimethoxyethane, Dichloromethane). The resolution was calculated against the closest peak.
Solution Stability
In this method, methanol was the first peak eluted, 2-butanone and ethyl acetate was the worst case for resolution, and 1, 2-dimethoxyethane presented the lowest S/N ratio. A mixture solution of these six solvents (SST solution) was prepared for system check and its stability was estimated (mixture at reference level) together with internal standard solution in well-sealed glass both at room temperature and in the refrigerator. The concentration of the solvent was checked by a freshly prepared solution employed as the external standard. The difference between the concentrations of the stored and fresh solution was calculated for review and assessment of the stability. The internal standard solution and SST mixture solution were stable for at least 200 days both at room temperature and under 2–8 °C in the refrigerator.
Accuracy:
The accuracy of the RRF method was demonstrated by testing samples where the solvent content is known by determination of external standard methods. In total, 20 laboratory samples containing different residual solvents in the validated linearity range were analyzed and the variation (relative difference) for all the tested solvents was below 10% with no matrix effects observed. The SST check is also evidence of accuracy in a sense since the recovery of internal standard (peak area of decane between blank and sample) can be obtained during carryover check.
Robustness:
The robustness of the method was evaluated by the experiment design with five centre changes of initial column temperature, ramp rate, flow rate, injection time, and equilibration time. The RRF value, resolution, and peak asymmetry were evaluated. The results showed that the method is sufficiently robust and there is no significant variation in the results in the operating ranges assessed.
In addition, the influence of the carrier gas and makeup gas type was evaluated in different laboratories using identical equipment and method conditions during method development. Hydrogen (H2), helium (He), and nitrogen (N2) were combined as carrier gas and makeup gas in three groups. The concordance of RRF value was checked, and deviation was within 10%. As demonstrated from the experiment, the influence of carrier gas and makeup gas was very limited on the RRF value and an easy method transfer between laboratories using different carrier gases could be foreseen.
Application and Advantages: The developed method was firstly applied in a non-GMP laboratory for process supporting and finally implemented in a GMP laboratory for in-process monitoring and controls for QC analyses after method validation. In 2017, about 400 samples were analyzed in a non-GMP laboratory using this method with dedicated HS-GC equipment (same brand
and model). Based on a 60-min saving for one sample (as compared in Figure 1 and Figure 2), about 50 working days were saved last year. In addition, with the HS-GC RRF method, only 5% of the sample diluent (NMP) was consumed compared with the previous year using traditional external standard method. This method is available for simultaneous determination of 25 residual solvents using just one sample injection, which means it is also a good choice to identify unexpected peaks and determine
the content without extra operation.
Conclusions
A simple, efficient, and robust method based on HS-GC for the simultaneous determination of 25 residual solvents has been developed and implemented in routine laboratories. It is sufficiently specific, linear, accurate, precise, sensitive, robust, and suitable as a quick and economic approach for the determination of residual solvents in a range of pharmaceutical substances, from process controls to product quality checks.
References
- International Conference on Harmonization, ICH Q3C (R6), Impurities: Guideline for residual solvents, current step 4 version, (ICH, Geneva, Switzerland, 2016).
- General Chapter <467> “Residual Solvents,” in United States Pharmacopeia 37–National Formulary 32 (United States Pharmacopeial Convention, Rockville, Maryland, USA, 2015), pp. 215–227.
- K. Rome and A. McIntyre, Chromatography Today May/June, 52–56 (2012).
- M. Tankiewicz, J. NamieÅnik, and W. Sawicki, Trends in Analytical Chemistry 80, 328–344 (2016).
- C. Cheng, S. Liua, B.J. Mueller, and Z. Yan, Journal of Chromatography A 1217, 6413–6421 (2010).
- K. Grodowska and A. Parczewski, Acta Pol. Pharm. 67, 13–26 (2010).
- S. Klick and A. Skold, J. Pharm. Biomed. Anal. 36, 401–409 (2004).
- R. Otero, G. Carrera, J.F. Dulsat, and J.L. Fabregas, Journal of Chromatography A 1057, 193–201 (2004).
Wenya Zhu is an analytical scientist in Chemical and Analytical Development, TRD, Novartis, Changshu, China. She studied analytical chemistry at university and has been involved in pharmaceutical analysis for over four years after graduation. She is experienced in LC and GC techniques.
Guoyi Liang is an analytical analyst in Chemical and Analytical Development, TRD, Novartis. He has 10 years’ experience in analytical science, and has special interest in GC techniques.
Wenlong Qiu is currently analytical technical lead in Global Product Development China, AstraZeneca. He previously worked at Chemical and Analytical Development China, Novartis, for 8 years before his current role. His special interests are LC and GC, method development strategies, and analytical control strategies.
Adrian Clarke is the analytical network leader in technical R&D, Novartis, Basel, Switzerland, and has responsibility for identifying the department’s strategic direction and technological needs in analytical chemistry. His special interests are liquid-phase separations (LC, 2D LC, SFC) and GC, method development strategies, and also analytical control and regulatory strategies.
Christoph Kolarik is an analytical scientist in Chemical and Analytical Development, TRD, Novartis, Basel, Switzerland. He has 35 years’ expertise in GC and hyphenated techniques, sample preparation, and traceâanalytics.
Sebastiano Mozzo has 21 years’ experience in the pharmaceutical industry at different levels and functions. Currently acting as a senior project manager in Lonza, the Netherlands, he is responsible for driving cell and gene therapy lateâphase projects through technology transfer and commercial production. Sebastiano previously worked at Novartis, Changshu, China, for over three years, where he was acting as analytical expert and development coordinator for the analytical R&D team.







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

