Ion Chromatography: An Overview and Recent Developments
LCGC North America
The basics of IC are reviewed and recent advances that may extend this technology to new applications are discussed.
The term ion chromatography (IC) was first used in 1975 to describe the chromatographic determination of inorganic anions using an anion-exchange separation and conductivity detection (1). This development allowed analysts to simultaneously determine inorganic anions such as fluoride, chloride, nitrate, phosphate, and sulfate in a convenient chromatographic format, replacing the tedious, expensive, and often inaccurate wet chemical assays. IC has been used in industries as diverse as environmental, power, semiconductor, electronics, and pharmaceuticals for a wide range of applications. Common applications include testing for common anions (U.S. EPA Method 314.1) and disinfectant by-products, such as bromate (U.S. EPA 302.0) in tap or bottled water, perchlorate (U.S. EPA Method 332.0, 314.2) and haloacetic acids (U.S. EPA Method 557) in drinking water, iodide and iodate in sea water, amines in wastewater, carbohydrates in fermentation broths, trace-level anions and cations in ultrapure water for the semiconductor and power industries, and more. In the pharmaceutical industry, IC is used throughout the manufacturing and regulation of pharmaceutical products, including the characterization of active ingredients, excipients, degradation products, impurities, and process streams (2). Sample types include raw materials, intermediates (including media and culture broths), bulk active ingredients, diluents, formulated products, production equipment cleaning solutions, and waste streams. IC is especially valuable for the determination of ionic or ionizable (in the mobile phase) analytes that do not have UV chromophores and therefore, by extension, combinations of analytes using multiple detectors in series.



Michael Swartz
Due to space limitations, a comprehensive treatment of IC is of course outside the scope of this column. While references are available that can provide additional details on IC, I'll try to cover at least some of the basics here (3,4). An IC instrument is a type of high performance liquid chromatograph, with slight differences in configuration and components. A schematic of a typical modern IC system is illustrated in Figure 1. It has some of the same basic components as a high performance liquid chromatography (HPLC) system: an autosampler, a high-pressure pump, an injector, an analytical column and guard column, a detector, and a data system. It also has some optional components not usually found in an HPLC system, including a suppressor or other form of a postcolumn reactor for derivatizations, and other forms of eluent generation and treatment. Because of the often corrosive nature of the mobile phase, the system components in contact with the mobile phase and the sample are typically made from inert materials, such as polyetheretherketone (PEEK). Traditional IC systems use chemical suppression: after the analytical column, the effluent is passed through a "suppressor" device that chemically reduces the ionic eluent background conductance (and therefore noise), while at the same time increasing the electrical conductance of the analyte ions (and therefore signal). An alternative separation using low capacity ion-exchange resins and low ionic strength eluants sometimes referred to as nonsuppressed IC, first reported in 1979, has also been used successfully for many types of analyses (5). Coupling the ion-exchange separation with any one of a number of detectors, for example, conductivity, UV, mass spectrometry (MS), inductively coupled plasma (ICP), and pulsed amperometric detection (PAD), expands IC applications to increased sensitivity and specificity. Because IC typically uses dilute acids, alkalis, or salt solutions as opposed to organic solvents in the mobile phase, reagent and disposal costs are more economical.

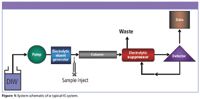
Figure 1: System schematic of a typical IC system.
Separation mechanisms for IC are primarily based upon ion exchange, although ion exclusion and ion-pairing approaches also are used. Selectivity is created by differences in charge density, hydration, size, and so forth of the ion-exchange site and polymer backbone, and on the valence, size, and polarizability of the individual ionic species to be measured. In some cases, reversed-phase separations are also performed on the basis of differences in the hydrophobic character of the ionic species.
Stationary and Mobile Phases
Since its inception in the early 1970s, progress in IC stationary phase development including the base material, the ligands or latex attached, the capacity, and smaller particle sizes has helped to meet new analytical challenges and maintain its popularity. An increasing number of column materials have been developed for IC, often targeted at specific analytical problems, aided by a better understanding of the various separation mechanisms. High speed, monolithic, and capillary columns have also been recently introduced (6,7).
Several different separation modes are used in IC including ion exchange, ion exclusion, and ion pair–reversed phase. Retention in ion-exchange processes is governed by the interaction between the mobile phase and ion-exchange groups bonded to the column stationary phase base material. Ion-exchange stationary phases traditionally consist of polystyrene, ethylvinylbenzene, or methacrylate resins copolymerized with divinylbenzene and modified with ion-exchange groups. Used for the separation of both inorganic and organic anions and cations, anion exchange usually is accomplished with quaternary ammonium groups attached to the polymer, whereas sulfonate, carboxyl, or phosphonate groups are used as ion-exchange sites for the separation of cations. Typically, anion separations use dilute bases, most commonly hydroxide or carbonate, as mobile phases on polymer-based anion-exchange stationary phases. Polymeric phases are more stable to extremes in pH compared to silica-based materials, and in many cases are still compatible with organic solvents. Silica-based columns that exhibit significantly higher efficiency can be used for cation separations, however, as dilute acids often serve as mobile phases and stability is not as much of a concern. Hydroxide is an eluting ion of choice because it is neutralized to water by the suppressor device. In cation analysis the acid eluent, most commonly methanesulfonic acid, is neutralized to water.
Just as with HPLC, mobile phases historically have been manually, and often painstakingly, prepared. Inconsistent mobile phase concentration as well as the presence of contaminating ions — for example the presence of carbonate in a hydroxide eluent — introduces retention time variability. However, with the development of electrolytic eluent generation, commonly referred to as reagent-free ion chromatography (RFIC), mobile phases can be prepared online. In this process, illustrated in Figure 2, an electrolytic eluent generator (EG) is placed inline between the high-pressure pump and the injection valve. Eluent ions are transferred electrolytically across a membrane from a reservoir containing a high concentration of ions into a stream of deionized water in response to an applied current. The resulting eluting ion concentration has greater accuracy, greater precision, and is easier to prepare than eluents made by traditional bench techniques. Because the hydroxide is produced online, there is no possibility of contamination from carbonate. This device also greatly facilitates gradient IC separations because it is more accurate than a pump-proportioning valve, required with manually prepared eluents.

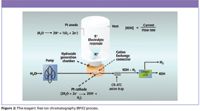
Figure 2: The reagent free ion chromatography (RFIC) process.
The composition and structure of the ion-exchange sites of the resins often are designed for certain critical separations. Other mechanisms can be added to enhance selectivity of certain target analytes, such as chelation or other complex formation. Fundamentally, retention increases with increasing ion-exchange capacity of the resin and valency of the analyte and decreases with inreasing ionic strength of the mobile phase and valency of the mobile phase. Other thermodynamic properties are also important in the design of the stationary phase and selection of the eluting ion.
The ion-exclusion IC separation mode is governed by ion exclusion, sorption processes, and, depending upon the column type, hydrogen bonding. Most commonly, a high-capacity, totally sulfonated cation-exchange chemistry based upon polystyrene–divinylbenzene is used as the stationary phase. Ion-exclusion chromatography is particularly useful for the separation of weak inorganic and organic acids from completely dissociated acids that are eluted as one peak within the void volume of the column. Ion-exclusion mobile phases generally consist of mineral acids when a UV or amperometric detector is used, although fluorinated organic acid eluents have been used for suppressed ion-exclusion chromatography.
The primary retention mechanism for the ion-pair mode of IC is adsorption. Analytes are separated based upon the ability to pair with a surface-active counter ion rather than based upon the ion exchange selectivity. In ion-pair IC the stationary phase consists of a neutral porous polymeric resin of low polarity and high specific surface area such as divinylbenzene, or in some cases, a traditional HPLC-type C18 stationary phase can be used. The mobile phase controls the selectivity. In addition to an organic modifier, an ion-pair reagent is added to the mobile phase taking into account the chemical nature of the analytes. Selectivity is complementary to ion-exchange selectivity rather than a substitute. Ion-pair chromatography is particularly suited for the separation of surface-active anions and cations, sulfur compounds, amines, and transition metal complexes but generally exhibits poor selectivity for polyvalent species.
IC Detection
Generally, the nature of the analyte dictates the mobile phase, column choice, and the detection mode used.
Conductivity detection is by far the most commonly employed mode of detection in IC. The conductivity detector is a bulk property detector that measures the conductivity of the mobile phase, and is the detector of choice for ion chromatography when the analyte does not have a UV chromophore (3,4). In a conductivity detector, the resistance (or strictly the impedance) between two electrodes in the flow cell is measured. Therefore, in IC, in which conductive buffers are required in the mobile phase, a suppressor column is placed into the flow path after the analytical column and before the detector to reduce the background conductance of the mobile phase. Termed "suppressed IC," the conductance of the mobile phase, often referred to as the background conductance, is reduced significantly as it flows through the suppression device. The suppression device is an ion exchanger that exchanges out or neutralizes the counterions of the eluent ion, that is, K+ from KOH, leaving only the analytes (and other co-ions) as the major conducting species in the effluent. The suppression ion in this case is H+ that can be supplied by a strong acid or, now more commonly, from the electrolysis of water. The reduced background conductance and the enhanced signal due to the ionic species in acid form result in an enhanced signal-to-noise ratio. Modern systems have incorporated several versions of continuously regenerated membrane-based suppressors (8), enabling the use of higher-capacity ion-exchange columns and gradient elution for the separation of mono-, di-, and trivalent acids.
The self-regenerating suppressor module relies on the return of the high-purity detector effluent and electrolysis to regenerate the suppressor. This simplification based upon recycling the water content of the mobile phase for use in the electrolytic generation of the regenerant acid (or base) eliminates the need for suppressor regeneration solutions (which can be expensive for cation IC), making IC viable for a wider range of applications.
Suppressed IC, while popular, does have some potential disadvantages. If a packed resin bed is used as a suppressor column, the suppressor column itself can contribute to peak broadening, decreasing efficiency, and also requires frequent regeneration. It should be noted that because conductivity detection requires that the analyte carry a charge at the pH of detection, very weak acids and bases can have poor detection limits or are not detectable at all.
Nonsuppressed IC, using low-capacity ion-exchange resins and low-conductivity eluents, allows conductivity detection with ion exchange–based separations and can provide some advantages. While suppressor-based methodologies must be implemented on systems specifically designed for IC, nonsuppressed IC can be performed without the suppressor on traditional HPLC systems, because the commonly used eluents in nonsuppressed IC include buffer systems that are compatible for use on existing HPLC instruments.
Mobile-phase components typically used in nonsuppressed IC are phthalic acid and p-hydroxybenzoic acid–based for the determination of anions, because the equivalent conductance values of chloride, sulfate, and other common anions are significantly greater than that of the eluent anion. For the determination of cations, the use of a methanesulfonic acid–based mobile phase is typical as the equivalent conductance values of sodium, potassium, calcium, magnesium, and other common cations are significantly lower than that of the cation (H+ ) in the eluent, and response is registered in the form of a negative peak. Nonsuppressed IC is easy to perform and useful for determining ions of weak acids such as cyanide and sulfide, which are nonconductive after chemical suppression, although electrochemistry can provide more useful detection limits.
PAD also is used commonly in IC. PAD uses a specialized mode of conventional amperometric electrochemical technology and is commonly used for the detection of electroactive species — for example, organic compounds such as carbohydrates, sugar alcohols, amino acids, and organic sulfur species. In PAD, the electrical potential is cycled several times a second. Its purpose is to clean and restore the detector electrode surface before a measurement is taken to generate the signal output. Analytes are detected by an oxidative desorption process at the surface of an electrode. The most common application is the determination of mono- and disaccharides on a gold electrode at high pH. The carbohydrates have pKa values in the 11–13 range, so very high eluent and stationary phase pHs are needed for an anion-exchange separation to operate. The USP recently added a method using high-performance anion-exchange with PAD (HPAE–PAD) to identify and quantitate the level of organic impurities in heparin due to recent health concerns (9–12). The organic impurities section of the new heparin USP monograph relies on hydrolyzing the polysaccharide and determining the relative amounts of galactosamine and glucosamine in the sample digests by analysis on an anion-exchange column with electrochemical detection. Heparin is composed of glucosamine and uronic acid and acid hydrolysis of heparin samples releases the glucosamine, which is determined readily with high sensitivity by pulsed amperometric electrochemical detection. In comparison, over-sulfated chondroitin sulfate (OSCS-the adulterant) is composed of galactosamine and uronic acid. In these compounds, acid hydrolysis releases galactosamine, also easily determined by electrochemical detection. The USP method measures the ratio of galactosamine/glucosamine as an indication of the heparin purity and to identify heparin samples that might be contaminated or adulterated with chondroitin sulfate compounds. The combination of the anion-exchange separation and the sensitivity of PAD allows for the detection of galactosamine at levels as low as 0.4% (11).
UV detection also is used commonly in IC for inorganic and organic ions that possess a UV chromophore. These include organic acids, bromide, iodide, nitrate, nitrite, thiosulfate, and cyano-metal complexes. UV detection may also be performed indirectly — for example in the analysis of cations, called indirect photometric detection. Direct photometric detection also can be used; it involves chelation of the metal ions in column effluent with a color-forming reagent before detection with a visible wavelength. A classic example is the separation of metal ions in which the column effluent is chelated with 4-(2-pyridylazo)-resorcinol followed by detection at 510–530 nm.
Another approach to IC based upon hydrophilic interaction liquid chromatography (HILIC), uses very polar stationary phases such as diol (neutral), silica (charged), amino (charged), or zwitterionic (charged) columns. The mobile phase is highly organic but contains a small amount of aqueous-polar solvent. This establishes a stagnant enriched water layer around the polar stationary phase allowing analytes to partition between the two phases based upon polarity. Water or a polar solvent is the strong eluting solvent. With zwitterionic columns and chromatographic conditions, the retention is due to a salt-exchange process simultaneously involving both the anion and cation. Using a zwitterionic stationary phase operating in the HILIC mode, the separation and quantitation of 33 commonly used pharmaceutical counterions, 12 cations, and 21 anions was reported (13). This work used evaporative light scattering detection (ELSD); however, corona charged aerosol detection (CAD) can better provide the high sensitivity, dynamic range, precision, and robustness desirable from a workhorse instrument in these types of critical pharmaceutical applications (14,15). Figure 3 shows a further advantage of the HILIC CAD combination; simultaneous analysis of several anions and cations in a single run, on a single system using one mobile phase, and one column.


Figure 3
Sample Preparation
Typical sample preparation for IC includes dilution or filtering through a 0.45-µm filter, or both. The most common form of sample preparation in IC is matrix elimination, wherein an interfering matrix ion or class of ions is removed while the analyte ions pass through unaffected. In a sense, this technique is based upon solid-phase extraction (SPE) techniques as the extracted species are matrix ions rather than analytes to be concentrated. For example, a highly alkaline sample can be neutralized by having it pass through a sample preparation cartridge packed with cation-exchange material in the acidic form. In this way, sample preparation cartridges can remove interfering matrices such as cations, anions, or hydrophobic substances that are encountered in many IC applications. The cartridge even can be installed inline between the autosampler and the injection valve, facilitating immediate, automated sample pretreatment. After being loaded into the injection valve sample loop, samples are processed automatically as they are eluted from one or more cartridges onto a concentrator column. Automated in-line sample pretreatment eliminates manual sample pretreatment steps, facilitates better separations, extends column life, and helps achieve reproducible parts per million–level determinations.
Newer IC System Technology
For a variety of reasons (speed, sensitivity, efficiency, resolution, and sample limitations) recent trends in LC technology in general have been towards smaller particles, and nano and capillary systems. IC is no exception. The first IC system supporting both analytical-scale (2–4 mm i.d. column) as well as capillary-scale (0.4 mm i.d. column) separations was introduced in early 2010 (16). When configured as a dual analytical and capillary channel system, analysts can operate one channel with analytical- scale columns while exploring capillary-scale separations on the other channel. These systems can support both manually prepared and electrolytic eluent generation on both analytical and capillary scales with eluent regeneration available when using 2–4mm i.d. columns.
Capillary-scale IC offers some real advantages over analytical-scale separations, as illustrated in Table I. Because of the very low flow rates, capillary scale IC can run continuously for several months on only 2 L of water. Less eluent consumption means less time spent making up eluents and equilibrating and calibrating the system. It also means that the system can be left on for extended periods of time and will be ready to perform analyses at a moment's notice. Also, the waste generated is much less than with analytical-scale systems, making capillary IC especially useful when analyzing toxic samples. Furthermore the injection volume is reduced to less than 0.5 µL while maintaining the same relative minimum detection limits, making capillary IC well suited for applications which are sample limited such as those found in metabolomics, monitoring individual corrosion spots and the process monitoring of small scale reactors.

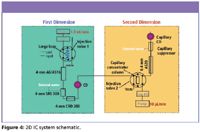
Figure 4: 2D IC system schematic.
For trace-level IC analyses, the fact that a capillary system requires 100 times less sample volume than an analytical-scale system means that only 1 mL of sample is loaded onto the concentrator column instead of the more typical 100 mL of sample needed with analytical-scale methods, resulting in substantial time savings.

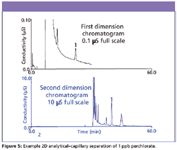
Figure 5: Example 2D analyticalâcapillary separation of 1 ppb perchlorate.
A dual channel system also offers the possibility of employing two-dimensional (2D) IC, or IC × IC, to provide greater sensitivity. In this configuration, as shown in Figure 4, high concentration interferences can be precisely removed from the sample in the first IC stage with low concentration analytes of interest retained and separated in the second IC stage. An example 2D analytical–capillary separation is shown in Figure 5. The time window from 19 to 24 min of the first dimension was loaded onto the concentrator of the capillary system in the second dimension, allowing a 100-fold improvement in detection response. Sensitivity also is enhanced through the use of capillary IC. An example of ultralow-level perchlorate detection is presented in Figure 6. Here, 30-ppt perchlorate is determined in bottled water using the 2D method.

Figure 6: Example capillary IC analysis of perchlorate at ultralow levels.
One of the keys to increasing sensitivity on capillary IC systems is a specially designed electrolytic eluent generator module that produces high-purity eluents from deionized water that yields low noise and stable baselines. The capillary format also increases the maximum generated eluent concentration to 200 mM, which can be used to further accelerate difficult separations. Using only 15 mL of eluent per day enables the system to be operated continuously. A capillary eluent generation cartridge will last up to 18 months under typical conditions.

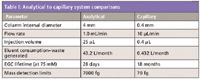
Table I: Analytical to capillary system comparisons
Capillary trap columns are high-pressure, electrolytically regenerated devices designed for use with hydroxide and methanesulfonic acid eluent generators. These trap columns operate continuously to remove trace levels of contaminants introduced by feed water and reduce baseline drift during gradient separations and improving day-to-day and laboratory-to-laboratory reproducibility. The capillary electrolytic suppressors are optimized for eluent flow rates typically used in capillary scale systems (5–30 µL/min).
Carbonate contamination from samples can decrease detection sensitivity and accuracy when measuring analytes like sulfate, which are eluted fairly close to the carbonate peak. Therefore, capillary systems typically use a carbonate-permeable membrane device to remove excess carbonate from the sample. This membrane is the same type as the ones used in standard and microbore devices but the dead-volume has been optimized for capillary-scale flow rates.
The general perception in liquid chromatography is that systems using smaller internal diameter columns — especially nano- and capillary columns — are much more difficult to use. True, operators must take great care when making fluidic connections and ensure that they are free of dead volume. To ensure the integrity of these connections, devices are now available that are designed to contain all capillary consumables in a single easily replaceable compartment. These devices house capillary and guard columns, the eluent generation degasser module, a loop injection valve, and the capillary electrolytic suppressor, reducing the number of fluidic connections by 50% compared to a similar analytical configuration. All tubing is precut and preformed, making capillary connections as easy and reliable as those for 4-mm i.d. columns.
Conclusion
Ion chromatography as an analytical tool continues to expand both in capability and application. New column chemistries, column formats, sample preparation devices and detection schemes continue to keep IC at the forefront of analytical laboratory technology. Newer capillary-scale IC technology offers improvements in speed, mass sensitivity, and economy in a system that can operate uninterrupted for up to nine months on a single 4-L volume of mobile phase, increasing operational convenience and reducing waste generation by 100-fold; a chemistry and technology that are both economically and environmentally "green."
Acknowledgment
The author would like to thank Phillip DeLand and Rosanne Slingsby of Dionex Corporation for contributions to this manuscript.
Michael Swartz "Innovations in HPLC" Editor Michael E. Swartz is Research Director at Synomics Pharmaceutical Services, Wareham, Massachusetts, and a member of LCGC's editorial advisory board. Direct correspondence about this column to "Innovations in HPLC," at lcgcedit@lcgcmag.com.
References
(1) H. Small, T.S. Stevens, and W.C. Bauman, Anal. Chem.47, 1801–1809 (1975).
(2) USP 33/NF 28, 2010, Chapter 1065.
(3) J.S. Fritz and D.T. Gjerde, Ion Chromatography, 4th Ed. (WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009).
(4) J. Weiss, Handbook of Ion Chromatography (Wiley, New York, 2005).
(5) D.T. Gjerde, J.S. Fritz, and G. Schmuckler, J. Chromatogr. 186, 509 (1979).
(6) R. Majors, LCGC 28(3), 192 (2010).
(7) R. Majors, LCGC 28(4), 290 (2010).
(8) J. Stillian, LC Mag.3, 802 (1985).
(9) Heparin Sodium monograph in The United States Pharmacopeia 32, Supplement 2, NF 25; American Pharmaceutical Association, Washington, DC, 2009.
(10) Heparin Sodium monograph in The United States Pharmacopeia 32, Supplement 2, NF 25; American Pharmaceutical Association, Washington, DC, 2009.
(11) Heparin Sodium, Pharmacopeial Forum 2009, 35(2), 1–10.
(12) Determination of Galactosamine Containing Organic Impurities in Heparin by HPAE-PAD Using the CarboPac PA20 Column, Application Note 233, LPN 2286, Dionex Corp., Sunnyvale, California, 2009.
(13) M.E. Swartz, LCGC 28(1), 53 (2010).
(14) D.S. Risley and B.W. Pack, LCGC 24(8), 776 (2006).
(15) M. Swartz, M. Emanuele, A. Awad, and D. Hartley, Advances in HPLC Systems Technology, Special Supplement to LCGC, 40 (April 2009).
(16) M.E. Swartz, J. Liq. Chrom., in press.
(17) M.E. Swartz, LCGC, in press.














. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

