Instrument Considerations in the Transfer of Chromatographic Methods, Part I: Method Considerations
LCGC North America
In the first of a three part series, we discuss challenges associated with successful transfer of chromatographic methods and how to best circumvent and remove ambiguities and contradictions
Successful transfer of chromatographic methods often proves more challenging than expected. This three-part article series will summarize the details of this process with particular attention to the sources of confusion and to some suggested solutions for the observed difficulties. In part I, the focus will be on the method as a whole and considerations for avoiding and eliminating ambiguities and inconsistencies between laboratories. In part II, the characterization of the system will be emphasized. In part III, emphasis will be placed the techniques for bringing the modules of the two systems into alignment.
Liquid chromatography (LC), in its instrumental form, is among the most common tools used for chemical analysis. The technique is fundamental in research, product development, quality control, natural product analysis and purification, and clinical testing across a wide range of application areas. Much effort and time is devoted to developing chromatographic methods for routine use. These methods must reliably separate each potential sample component in a way that is suitable for unequivocal identification and for quantitative analysis. These well-developed methods become part of standard operating procedures and, ultimately, regulatory documents, as well as being very desirable for use in related laboratories. This transfer of methods has proven generally challenging in many cases. This is a very large topic, and we will focus this discussion on some subsets of the general problem.
Method transfer can be attempted for several reasons. A laboratory may try to implement an established method when working on the same or a similar analytical problem. This process tends to be fairly simple since the original method is only a guidance to a starting point. At the other extreme, a laboratory may be implementing a method to obtain the exact same result as the originating laboratory. This approach can occur within an organization or company because of a need for expanded capacity, or to analyze the same sample types at a new location. In the latter case, it is planned to duplicate exactly the chromatogram obtained in the originating laboratory as documented in a standard operating procedure or regulatory document. In this case, the same qualitative and quantitative analytical results are required. Between these extremes, the range of possible objectives includes a desire to improve a method in any of several ways, including modernizing the materials and instrumentation, reducing run time or operating cost, improving sensitivity or accuracy, or adding validation for additional analytes like newly recognized impurities. For this discussion, we will focus on the most rigorous method transfer leading to the duplication of results of the established methods without improvements to the method. The considerations in this approach also apply to the other forms of transfer, with some additional factors to be considered elsewhere. We will assume that the methods under consideration include detailed materials and methods, system suitability criteria, and expected results with acceptable limits. We will also include consideration of the many things that can go wrong in such an exercise.
This discussion will be divided into three parts. In this first part, we will consider those aspects of the method itself that affect the transfer of the method. In the future, part II will address the chromatographic systems, and part III will consider the details of aligning individual instrument modules.
General Considerations for Effective Method Transfer
A consistent principle in the discussion to follow is the elimination of all sources of variability, particularly in the early stages of the method transfer. The use of identical materials and methods creates a stable point for any further use. After the method is operating as specified, modifications and substitutions can be made in a controlled way. Although this principle seems obvious, it is often not observed in practice. At the same time, as we will emphasize below, it is assumed that the originator of the method followed all the same principles and controls in developing the method. Uncontrolled and undocumented elements contribute to a method that is difficult or impossible to transfer.
The elements that must be considered in a successful method transfer include the procedures, the sample and its preparation, the available chemicals, the ancillary laboratory instruments and equipment, the chromatographic column, the mobile phase, and the chromatographic instrument itself. In addition, the skills and experience of the laboratory scientist or operator are often overlooked variables in the process.
Even highly skilled laboratory personnel differ in their knowledge and in the exact way that they do particular tasks. If at all possible, it is desirable to establish communication between the new operator and either the developer of the method or a person who has routinely and successfully executed the method. The consultation should begin at the start of the process, and be available until successful transfer is accepted. If such collaboration is not possible, it would be useful for two people to share the transfer exercise to allow for different experiences and consideration of different details in execution of procedures.
The written procedures for the established method are expected to provide sufficient information and detail for reasonably trained and skilled scientist. Every individual who prepares such a document tries to meet these criteria. We must, however, recognize that each of us who writes such procedures is affected by the things that they know and by the details that they recognize without additional thought. We all omit writing down the things that "everybody knows." Our intended audiences do, however, know somewhat different things. We should remember for example that there are at least four reasonable ways to prepare 50:50 methanol–water, as discussed below. The same is true of a buffer, such as 25 mM sodium phosphate, pH 7, also with four different recipes. An example of the differences in the separation resulting from different preparation protocols is shown in Figure 1. The differences in retention are small, but they exceed the width of the peak. Each detail must be described as explicitly as possible to ensure that it will be faithfully replicated in the new adopter laboratory. This guideline becomes more important each year with increasing globalization. The receiving laboratory may not share the primary language of the originator. Incorporation of more detail, rather than less, is the best practice.


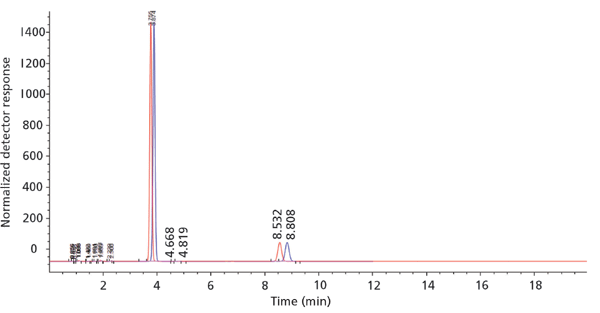
Figure 1: Effect of Solvent Preparation Protocol: Red trace - Mobile phase prepared on demand with instrument blending; Blue trace - Mobile phase prepared volumetrically by laboratory scientist.
The sample and its preparation can often prove the most challenging aspect of a method transfer. Because this is such a large and diverse topic, we will not consider it in this discussion. It is worth noting that the steps in sample preparation, including preparation of the standard, require the same level of detail and specificity as the other parts of the method.
The chemicals chosen for the transferred method will often be chosen based on those readily available in the laboratory or in a local stock room. This approach often proves unwise. The reagents used for standard and sample preparation may contribute chromatographic peaks or compromise the recovery of important analytes. The best practice is to duplicate the reagent set, not just grades but even the same chemical vendor where possible. Although it may seem unnecessary to purchase new supplies of common reagents, it is desirable for the first trials in a method transfer to eliminate as many variables as possible. After successful chromatographic analyses are obtained, it is possible to change reagents in a controlled way, confident that any consequences of reagent composition can be quickly recognized. This statement assumes that the originating laboratory used reagents that were relatively fresh and uncontaminated.
A variety of laboratory equipment, in addition to the liquid chromatograph, will be used in executing the method. This equipment includes instruments such as pH meters, balances, digital pipets. All these devices must be maintained in a reliable, calibrated state to be used in a successful method transfer. It should be obvious that this calibration and maintenance must be rigorous on both sides of the transfer. For example, incorporating an erroneously measured pH into a procedure can preclude successful use of the method by anyone else. In addition, it is commonly found that accessory instruments are not identical. Particularly with respect to sample preparation, the results can be different with some types of tools. This advice applies not so much to balances and pH meters as it does to homogenizers or centrifuges.
The Chromatographic Column
The chromatographic column is a frequent source of discrepancies between laboratories. Such difficulties should be easily avoided by restricting initial trials to the use of only the exact column specified in the method. The major column manufacturers have become quite rigorous in maintaining consistent properties within a particular brand of column of a given particle size and dimensions. Batch controls are good, and it is possible to obtain multiple columns from a single batch, as well as columns from a representative set of batches. Problems can arise when a different column is substituted for that specified in the method. In addition, difficulties have often arisen when a previously used column is chosen for a transferred method. This approach is extremely unwise because the history of a column is usually unknown and impossible to duplicate. Such prior use may leave some trace residue on the column surface, or otherwise modify the chemistry. As always, this principle can only be successful when the originator of the method established operating limits with a column with no history, and screened multiple columns from multiple batches.
Mobile-Phase Preparation
Preparation of the mobile phase has proven to generate more difficulties than one would anticipate. Such problems generally arise from either the quality of the mobile-phase components or from the preparation. The components of the mobile phase, both aqueous and organic solvents, can distort the baseline, contribute additional chromatographic peaks, or alter the selectivity of the separation. Water is an especially variable solvent, so either an on-demand laboratory water purification system or high-purity bottled water is safest. Organic solvents should be the highest possible quality, especially for the first test, and they must be at least the specification used in the originator trials. All solvent components, including any additives, should be freshly opened and prepared.
It is possible to prepare the mobile phase differently from the originator laboratory. As noted above, there are at least four reasonable ways to make 50:50 methanol–water: place 500 mL of water in a 1000-mL volumetric flask and bring to volume with methanol; place 500 mL of methanol in a 1000-mL volumetric flask and bring to volume with water; measure 500 mL of methanol in a graduated cylinder and 500 mL of water in a second graduated cylinder; and weigh 500 g of methanol and 500 g of water. These four formulations will give measurably different retention and chromatographic selectivity. There are even more ways to prepare 25 mM sodium phosphate, pH 7.00: titrate 25 mM phosphoric acid with concentrated sodium hydroxide; titrate 25 mM monobasic sodium phosphate with concentrated sodium hydroxide; titrate 25 mM dibasic sodium phosphate with phosphoric acid; blend solutions of 25 mM monobasic sodium phosphate and 25 mM dibasic sodium phosphate; mix specified volumes of 25 mM monobasic and dibasic sodium phosphate as calculated from published values for pKa or from commonly available tables in reference books; or weigh the amounts of solid monobasic and dibasic phosphate salts, again as calculated. These different formulations may give altered chromatography in reversed phase, especially for ionic analytes, and will certainly affect ion exchange chromatography. In addition, problems commonly arise from failure to observe the different formula weights associated with different hydration states of salts and from not correcting for the effects of temperature on pH measurement.
It would be best practice in developing and documenting a standard method to choose one of the alternatives described above and to write a detailed description of what was actually done. Gravimetric preparation of aqueous–organic mixtures is probably most exactly communicated, but allowance must be made for not every laboratory having a balance of sufficient capacity for accurately weighing the required amount of solvent, typically more than a kilogram.
An alternative approach to mobile-phase preparation has been suggested and is somewhat frequently used. It is possible to use a multisolvent chromatographic pumping system to blend pure solvents on demand. The desired percentages of each solvent are programmed into the gradient table or the isocratic pump control. This approach removes the manual preparation steps to reduce labor. It tends to be more reproducible than manual blending since fewer measurements are made. Although the technique generally gives accurate results, there are sources of imperfect results that are discussed in some depth below in the context of instrumental characteristics. Purely from the perspective of solvent blending, it would be good practice to compare a batch of preblended mobile phase with the results from an instrument-blend. If the results are both within the specified limits, the labor-saving technique can be implemented. Typical results are shown in Figure 1.
Instrumentation-General Considerations
The last topic to be considered, the chromatographic instrument itself, is also the largest. The common principle applied for all other considerations-use exactly what was used in the originator's laboratory-is desirable here. It is, however, very often impossible to maintain consistency. The instruments selected in various laboratories are often different models or brands, and it is not usually financially sensible to purchase chromatography instruments for each specific, new method to be implemented. Furthermore, the usable lifetime of a method is often much longer than that of an instrument. Matching an instrument, therefore, may not be possible to begin and execute a method transfer. We must, therefore, consider the differences among instruments that can affect method transfer. The transfer of a method from one instrument to another may require some adjustment of the method. Many laboratories adhere to the guidelines found in Chapter 621 of the current United States Pharmacopeia (USP) (1). The currently applicable chapter specifically states
“Adjustments to the specified chromatographic system may be necessary in order to meet system suitability requirements. Adjustments to chromatographic systems performed in order to comply with system suitability requirements are not to be made in order to compensate for column failure or system malfunctions. Adjustments are permitted only when . . . adjustments or column change yields a chromatogram that meets all the system suitability requirements specified in the official procedure.”(1)
These guidelines, often mentioned as "<621>," specify ranges of changes to the method that may be implemented without revalidating the method. The chapter has been summarized in many places, but the original document should always be consulted. We will allude to specific items in the context of specific challenges in method transfer. It should be emphasized that many laboratories follow these limits and practices, but they are not universal regulations. They are absolute requirements only for USP compendial methods.
Conclusions
We have considered, to this point, the characteristics of a method that can affect the transfer of a chromatographic method from one user or laboratory to another. Some suggestions have been included for ways to avoid difficulties. It has been noted that many of these difficulties are rooted in method descriptions that can be interpreted differently by well-trained scientists or executed in alternative ways by skilled laboratory workers. There has been an emphasis on providing detailed descriptions that can minimize such differences. In the next installment, the focus will shift to the instruments used for the origination and the execution of the chromatographic method.
References
(1) General Chapter <621> "Chromatography" in United States Pharmacopeia 40 National Formulary 35 (USP 40-NF 35, United States Pharmacopeial Convention, Rockville, Maryland, 2017), pp. 508–520.
Thomas E. Wheat is a principal scientist with Chromatographic Consulting, LLC in Hopedale, Massachusetts. Direct correspondence to: chromatographic.consulting@gmail.com.










; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")



. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.


