The Increasing Role of Superficially Porous Particles in HPLC
A review of the development of superficially porous particles and what the latest advances in this technology offer chromatographers in practice.
Among the first packing materials developed for high performance liquid chromatography (HPLC) in the late 1960s, the superficially porous particle (SPP) has been available for over 40 years in various forms. Most recently, this concept of a packing with a solid core and a thin porous shell has regained momentum, where sub-3 μm SPPs give rise to the efficiency of sub-2 μm porous particles yet have half of the pressure drop. This instalment of "Column Watch" will trace the development of SPP supports to the present-day versions (recently termed core shell, fused core and porous shell packings) that rival ultrahigh-pressure liquid chromatography (UHPLC) supports. Modern-day SPP columns can be used for the separation of both small and large molecules.
In most forms of classical liquid chromatography and high performance liquid chromatography (HPLC), totally porous particles such as silica gel, alumina and ion exchangers have been used in most modes of chromatography. These materials are useful because they present a high surface area, show good retention and sample capacity, and provide a base for the further bonding of organic moieties.
In the early 1960s, HPLC as we know it today hardly existed. Although researchers had predicted theoretically that liquid chromatography could be more efficient and faster, little practical work had been done to realize these predictions. At that time, most chromatographers used open glass columns packed with porous particles larger than 100 μm. Most of these particles were obtained by sorting wide size ranges of irregularly shaped particles into narrower cuts using sieves. Chromatographers used gravity as the pressure source to move liquids through the glass columns, collected analytes at the column exit, and measured the analytes in the effluent using off-line techniques such as UV–vis spectroscopy. By today's standards chromatographic peaks were very broad. Attempts to speed up the chromatography by applying positive pressure only resulted in broader peaks because of slow mass transfer into and out of these large porous particles. Something had to be done to speed up chromatography. Clearly for highspeed separations, the solute migration distance or pore depth had to be decreased or the rate of solute diffusion had to be increased.
Early SPPs
The theoretical work of Purnell,1 Golay2 and Knox3 and experimental GC results in Horvath's PhD thesis4,5 predicted that using a support with an impenetrable hard core coated with a thin outer layer of a porous solid should provide high-speed solute mass transfer in the stationary phase. Horvath and coworkers6 first demonstrated the so-called porous layer bead (PLB, also referred to as a pellicular or superficially porous particle, SPP) support for the ion-exchange chromatography of nucleotides. This SPP consisted of a glass bead solid core with a thin layer of polystyrene–divinylbenzene polymeric resin modified with anion-exchange groups on its surface. Figure 1 depicts how this new concept compared with the large porous particles of yesteryear. Figure 1(a) shows a typical large porous particle used in the early days. When a solute came down the column it had to diffuse into the pores of the particles (millions of times as it moved through the column) and because of this slow diffusion into and out of the stagnant mobile phase, a great deal of time is required. Meanwhile, the mobile phase that the solute was originally in has long since passed the particle. This slow mass transfer is a source of band broadening in chromatography. Of course, the temperature of the column could be increased to decrease mobile phase viscosity but this resulted in only minor improvement and at the time was inconvenient.


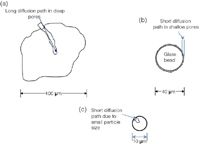
Figure 1: Comparison of solute mass transfer (migration) distances in (a) large porous particles with irregular shape; (b) early SPPs (~1970); and (c) small porous particles with spherical shape (mid-1970s).
Figure 1(b) depicts the concept of the SPP. A spherical solid glass bead, 40 μm in diameter, was used as the base material. The outside the glass bead was coated with a thin layer of ion-exchange resin,6 silica gel7 or controlled surface porosity beads.8 The layer was usually 1–2 μm thick. Now the solute would come down the column and diffuse into the SPP, but the diffusion distance was limited by the underlying solid core. By limiting the deep pores, the SPP diminished the pathlength for diffusion in the stagnant mobile phase. Diffusion could only occur in the thin layer on the surface so that rapid mass transfer took place. From a theoretical point of view, the thinner the layer the faster the mass transfer will be, and the better the efficiency. Of course, as the layer becomes thinner, both the capacity factor (retention) and the sample capacity (loading) of the SPP decreases so that its separation potential is less. Thus a compromise must be made between the layer thickness and the capacity.
Compared with the large, porous particles used earlier in LC, SPPs provided much faster separations (in the 20–30 min range, rather than several hours), more efficient separations (height equivalent to a theoretical plate, HETP, in the 1–2 mm range), and better limits of detection. Users could easily dry pack the dense, solid core pellicular packings into 50 cm and 1 m stainless steel columns using mechanical vibration. Many chemical bonded phases were developed to provide various separation mechanisms especially for normal-phase, reversed-phase and ion-exchange chromatography. Many successful applications using SPP columns were developed in the early 1970s.
The invention of commercial SPPs in the late 1960s and early 1970s, along with the advent of the flow-through UV detector were the two occurrences that kicked off the development of HPLC. Of course, the breakthroughs in the sizing and packing of small silica porous particles in the early 1970s9,10 that resulted in further efficiency increases led to the demise of large SPPs. The microparticulate packings [depicted in Figure 1(c)] improved mass transfer even more and improved efficiency by at least another order of magnitude. In addition, the high surface area of these small porous particles provided much greater sample capacity than SPPs and these packings could also be used for preparative work and with larger sample injections for better detection. Readers interested in the complete story can refer to an earlier publication I wrote of the subject of the commercial development of SPPs and microparticulate packings.11
Through the decades, newer versions of SPPs have occasionally reappeared to address alternate solutions to specific problems. Although the large particle SPPs were widely used in the 1970s they gave way to the microparticulate packings which took over usage in the 1980s and early 1990s and remain the favoured HPLC packing today. After the 1970s, the large-particle SPPs were relegated to previously validated methods or as an inexpensive, easily replaced packing material for guard columns.
Modern SPPs
Rather than a 40 μm diameter particle, the first modern SPPs were 5.0 μm in diameter with a 0.25 μm layer and 300 Å pores.12 Instead of a glass bead, solid silica cores were used as a substrate. Then, a thin film of silica nanoparticles was coated onto this core. These SPP columns were recommended for the rapid separation of biomacromolecules such as proteins.13 Large biomolecules have very low diffusion coefficients, about 1/10th that observed for a small molecule. The thin shell of the SPP allowed the slowly diffusing proteins to only partially penetrate the packing (since the solid core prevented further diffusion). Compared with totally porous particles of the same size, mass transfer was more rapid and a high flow-rate could be run without sacrificing much column efficiency.12 In Figures 2(a) and 2(b), 5 μm porous particles and 5 μm superficially porous particles are depicted. For the totally porous particle [Figure 2(a)], assuming that the solute molecule diffused to the centre of the particle and then back out again, the total solute migration distance in this particle would be 5 μm (2.5 μm in and 2.5 μm out). The large biomolecule will diffuse very slowly in this distance resulting in rather broad peaks, especially as the flow-rate was increased for a more rapid separation.

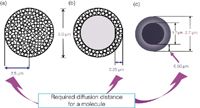
Figure 2: Comparison of diffusion distances in modern HPLC packings: (a) 5 μm totally porous particles; (b) 5 μm SPPs; and (c) 2.7 μm SPPs.
Contrast this to Figure 2(b) that depicts a bonded phase SPP of the same particle diameter. Assuming a protein will diffuse into and out of the thin layer, the total migration distance would be 0.5 μm (0.25 μm in and 0.25 μm out). This distance would be 1/10th that of the totally porous particle. Thus the diffusion time is much less and the peak width should be narrower. Even at higher flow-rates, the peak widths would be narrower than the porous particle at the same flow-rate.12
This comparison can be made by looking at the chromatograms of Figures 3(a) and 3(b). Here, for a couple of proteins, lysozyme and myoglobin, were separated at increasingly higher flow-rates on a 75 mm × 2.1 mm column packed with totally porous wide-pore (300 Å) bonded silica particle [Figure 3(a)] and a superficially porous wide-pore (300 Å) bonded silica particle [Figure 3(b)]. As is usual for proteins, the chromatograms were run under gradient conditions. Had the chromatograms been run under isocratic conditions, the differences in column efficiency would have been even greater. Under gradient conditions, the SPP column showed narrower peaks than the totally porous particle column as a result of more rapid mass transfer. One can see that the small peak on the leading edge of lysozyme remains partially resolved on the SPP column. Note that the chromatograms were aligned to make the comparisons easier. Also, as the flow-rate increased, the retention times on both columns decreased proportionally. Also, the gradient times had to be adjusted as the flow-rate was increased.


Figure 3: Comparison of totally porous particle and SPP columns at increasingly higher flow-rates for biomacromolecules under gradient conditions. (Chromatograms are aligned for comparison.) Columns: (a) 75 mm à 2.1 mm, 5 μm Zorbax Stablebond C18, totally porous particle; (b) 75 mm à 2.1 mm, 5 μm Poroshell 300 Stablebond C18, superficially porous particle; mobile phase A: 95% water, 5% acetonitrile with 0.1% trifluoroacetic acid; mobile phase B: 5% water, 95% acetonitrile with 0.07% trifluoroacetic acid; temperature: 70 °C; detection: UV at 215 nm. Peaks: 1 = lysozyme, 2 = myoglobin.
To illustrate the resolving power of a superficially porous wide-pore particle column, Figure 4 shows the rapid gradient separation of eight proteins in just over 0.75 min at a very high linear velocity for a 2.1 mm i.d. column.

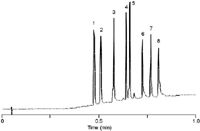
Figure 4: High flow-rates with 2.1 mm i.d. SPP column for high resolution and fast protein separations. Column: 75 mm à 2.1 mm, 5 μm Poroshell 300SB-C18; mobile phase A: 0.1% trifluoroacetic acid; mobile phase B: 0.07% trifluoroacetic acid in acetonitrile; gradient: 5â100% B in 1.0 min; flow-rate: 3.0 mL/min; temperature: 70 °C; pressure: 250 bar; detection: UV at 215 nm. Peaks: 1 = angiotensin II, 2 = neurotensin, 3 = RNAse, 4 = insulin, 5 = lysozyme, 6 = myoglobin, 7 = carbonic anhydrase, 8 = ovalbumin.
Sub-3 μm SPPs
Previous results show that the superficially porous 300 Å particles are suitable for the rapid separation of macromolecules. For small molecules, the recent development in sub-2 μm porous particles (1.5–1.9 μm) has allowed for high-speed separations. Indeed, short 50 mm columns packed with sub-2 μm particles give rise to separations in the minute range and longer columns (up to 150 mm) can generate tens of thousands of theoretical plates. However these columns can generate very high back pressure due to the small particle size, sometimes necessitating UHPLC systems.
Once again, only recently, the concept of SPPs was revisited. The new generation of SPPs, also called fused-core-, shell-, porous-shell and Poroshell particles, was introduced. These new SPPs have a particle diameter of 2.6–2.7 μm, but with a 0.5 μm porous layer and 90–120 Å pores are more suited to small molecule separations. These 2.6–2.7 μm SPPs provide a lower pressure drop (roughly 40–60% lower) than that observed for the sub-2 μm porous particles yet the short diffusion pathlength (slightly lower C term of the van Deemter equation) and other favourable particle characteristics give rapid solute mass transfer indicative of a sub-2 μm porous particle. Using a special synthesis process, very narrow particle size distributions of SPP can be achieved, much narrower than can be obtained with porous particles. Thus, these dense particles can be packed very efficiently into an HPLC column. This favourable particle configuration gives a lower A term (eddy diffusion) in the van Deemter equation. In addition, studies have reported a reduction in the B term.14 By a combination of the favourable lowering all of three terms in the van Deemter equation, efficiency of a column packed with these SPP particles rivals the sub-2 μm particles. Thus, high efficiency at lower pressure allows users to adapt their conventional LC systems without having to invest in UHPLC hardware.
On the other hand, having UHPLC hardware allows one to use very long SPP columns to generate high numbers of theoretical plates. Figure 5 compares two high-efficiency separations performed with SPPs and totally porous sub-2 μm particles.15 Most comparisons done nowadays are shown on short columns in high-throughput applications, usually with some minute time scales. This example shows very long columns that together generated about 100000 theoretical plates for both types of packings. Both sets of columns had 55 cm × 2.1 mm dimensions. In addition to the plate counts, the overall separation times were roughly the same. The major difference in the current chromatograms was the column pressure drop. For the totally porous sub-2 μm particle column, the pressure was just over 1000 bar (15000 psi), while for the 2.7 μm SPP column, the pressure was only 547 bar (8200 psi).


Figure 5: High efficiency separations on 2.7 μm SPP and totally porous particle sub-2 μm columns. Top: 55 cm à 2.1 mm (3 à 15 cm, 10 cm), 2.7 μm Poroshell 120 EC-C18 (SPP); mobile phase: 56:44 acetonitrileâwater; flow-rate: 0.202 mL/min; temperature: 40 °C; pressure: 547 bar. Bottom: 55 cm à 2.1 mm (3 à 15 cm, 10 cm), 1.8 μm Zorbax RRHD Eclipse Plus C18 (totally porous particle); mobile phase: 59:41 acetonitrileâwater; flow-rate: 0.23 mL/min; temperature: 40 °C; pressure: 1001 bar. (Reproduced with permission from reference 15.)
Commercial Availability of Modern SPP Columns
So far, there are only a few companies that have introduced superficially porous particles for macromolecular and small molecule separations (see Table 1). Besides a 2.6 μm particle size, Phenomenex (Torrance, California, USA) has introduced a 1.7 μm SPP that would have a similar pressure drop to the sub-2 μm UHPLC materials, but should offer even higher efficiency, perhaps stressing modern UHPLC instrumentation in terms of extracolumn contributions to band broadening. Because of the acceptance of SPPs for both HPLC and UHPLC, it is anticipated that additional companies will introduce products in the near future. In addition, more phases for the sub-3 μm SPPs are under development so that there will be as many chemistries available as for sub-2 μm and regular HPLC particles.

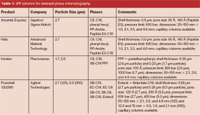
Table 1: SPP columns for reversed-phase chromatography.
Conclusions
The concept of SPP supports has been around a long time in LC and now has reemerged in a format that rivals separations achieved on small porous particles with sub-2 μm diameters. The newer SPP supports provide rapid separations of large biomolecules as well as small molecules, the latter with lower pressure drops than the sub-2 μm competitive columns. Although not discussed here, SPPs for small molecules have thicker porous layers and thus their sample capacity is not greatly reduced as was observed in early large-particle SPPs. One can do the calculation but the available surface area is only reduced 25% compared to a totally porous particle. However, the particle density for the SPPs is higher and therefore the amount of stationary phase packed into an SPP column is comparable to that packed into a totally porous particle column. Thus, the total sample capacities of the two types of columns are similar. Because the 2.6–2.7 μm SPP columns for small molecules and the 5.0 μm SPP columns for macromolecules have regular 2.0 μm porosity inlet and outlet frits, they are less prone to plug than the sub-2 μm particle columns that have smaller porosity frits. Clearly, there is a bright future for such packing materials as users become more familiar with the technology.
"Column Watch" editor, Ronald E. Majors, is a senior scientist at the Columns and Supplies Division. Agilent Technologies, Wilmington, Delaware, USA and is a member of LCGC Asia Pacific's editorial advisory board. Direct correspondence about this column should be addressed to "Column Watch", LCGC Asia Pacific, 4A Bridgegate Pavilion, Chester Business Park, Wrexham Road, Chester CH4 9QH, UK or e-mail amatheson@advanstar.com
References
1. H. Purnell, Nature, 184, 2009 (1959).
2. M.J.E. Golay, in Gas Chromatography 1960, R.P.W. Scott, Ed. (Butterworths, London, 1960), p. 139.
3. J.H. Knox and L. McLaren, Anal. Chem., 35, 449 (1963).
4. Cs.Horvath, PhD Thesis, Universitat Frankfurt am Main, Germany, 1963.
5. I. Halasz and Cs. Horvath, Anal. Chem., 36, 1178 (1964).
6. Cs. Horvath, B.A. Preiss and S.R. Lipsky, Anal. Chem., 39, 1422 (1967).
7. R.E. Majors, J. Chromatogr. Sci., 8, 338 (1970).
8. J.J.Kirkland, J. Chromatogr. Sci., 7, 361 (1969).
9. R.E. Majors, Anal. Chem., 44, 1722–1726 (1972).
10. J.J. Kirkland, J. Chromatogr. Sci., 10, 593–599 (1972).
11. R.E. Majors, LCGC North America, 12(7), 508–518 (1994).
12. J.J. Kirkland, Anal. Chem., 64, 1239–1245 (1992).
13. R. Ricker, C. Woodward and R.E. Majors, Amer. Lab., 36(14), 25–32 (July 2004).
14. F. Gritti and G. Guiochon, submitted for publication.
15. J. Link et al., "A Critical Performance Comparison of Column Options Using Poppe and Kinetic Plots", Pittcon 2010, March 2, 2010, paper 1230–4.

















Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.


Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.

