An Improved GC–MS Method for Cigarette Smoke Characterization Using a Novel Cold Trap, Dual Column, and Cryofocusing System
LCGC Europe
Cigarette smoke is a highly complex matrix and presents analytical difficulties for the analyst performing compound identification by gas chromatography analysis coupled with mass spectrometric detection (GC–MS). The development of a novel trapping system and a modified GC–MS layout (using dual chromatographic columns and cryogenic focusing devices) has improved the chromatographic separation of volatile and semi-volatile compounds found in cigarette smoke. This improvement has led to the potential to identify compounds usually masked by the solvent peak. This approach has also reduced the amount of peak overlapping by increasing the chromatographic peak capacity with the use of two capillary columns chosen for their analytical specificity.
Cigarette smoke is a highly complex matrix and presents analytical difficulties for the analyst performing compound identification by gas chromatography analysis coupled with mass spectrometric detection (GC–MS). The development of a novel trapping system and a modified GC–MS layout (using dual chromatographic columns and cryogenic focusing devices) has improved the chromatographic separation of volatile and semivolatile compounds found in cigarette smoke. This improvement has led to the potential to identify compounds usually masked by the solvent peak. This approach has also reduced the amount of peak overlapping by increasing the chromatographic peak capacity with the use of two capillary columns chosen for their analytical specificity.


Photo Credit: Cordula Damm/EyeEm/Getty Images

Cigarette smoke is a complex matrix containing between 4000 and 5300 different identified compounds (1,2,3). When performing gas chromatography–mass spectrometry (GC–MS) analysis on cigarette smoke extracts, scientists are usually confronted with two main difficulties: The first is the limited chromatographic space, which leads to unresolved peaks, and the second is the risk of chemical interactions between the sample and the collection solvent.
Cigarette smoke is known to consist of two distinct parts: the particulate phase, which can be trapped using a glass fibre pad filter (as described in ISO 4387 [4] and ISO 22634 [5]), and the volatile phase, which can be collected in sampling bags (6,7) or by impingers containing a trapping solvent either at room temperature (8) or cooled (9,10). A further trapping method has been detailed using a cryogenic instrument (11), with a liquid extraction after the smoking run. However, all of these trapping methods have drawbacks: the glass fibre trap does not allow the collection of volatile compounds; and the trapping in sampling bags leads to rapid decomposition of unstable species, which requires timeâdependent handling to avoid significant analytical variations. All of the described trapping methods using solvents result in sample dilution, which has the potential for chemical interactions between the trapped compounds and the solvent.
On the chromatographic side, the choice of column is a critical factor because the samples usually contain compounds that cover a wide range of volatility and polarity. Column selection is therefore usually a compromise between suitability and optimization for a large variety of compounds.
The objective of this project was firstly to develop a solventâfree trapping tool for cigarette smoke that is efficient and simple to use, and secondly to improve separation by using two different chromatographic columns.
A glassware device was developed to trap cigarette smoke at a low temperature in a vial compatible with the headspace sampler. The GC–MS instrument was modified to improve the chromatographic separation by the addition of two cryoâtraps, each preceding a chromatographic column of different polarity (adapted to the type of chemical compounds), which significantly increased the total peak capacity of the analysis.
Accordingly, several further technical modifications were applied to allow the simultaneous use of two chromatographic columns for the analysis of the samples, which led to the development of a new alternative analytical platform described in this article.
Experimental
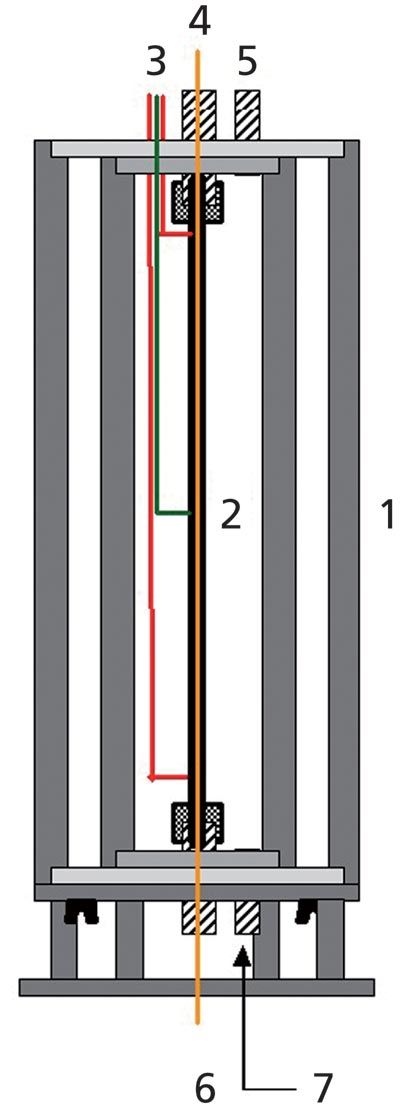
The reference cigarettes 3R4F were supplied by the University of Kentucky and conditioned according to the ISO 3402 standard (12) (at least 48 h at 60% relative humidity and 22 °C). They were inserted into the cigarette holder (Figure 1, part 8 and 9) and were lit with an electric lighter. Eight puffs were taken following the ISO 3308 standard (13) (one puff per minute, puff volume: 35 mL, puff duration: 2 s, puff shape: bell curve) using a puff generator developed in-house. The cigarette smoke was collected using two traps mounted sequentially (Figure 1, parts 2 and 8). The first one, a standard glass fibre filter according to the ISO 3308 standard (part 8 in Figure 1) (Borgwaldt), collected the particulate phase of the aerosol. The second trap, a custom-made glass vial (Figure 2) cooled to a temperature of -150 °C using a flow of liquid nitrogen regulated by an electronic controller, collected the volatile compounds representing the vapour phase.

Figure 1: HS-cold trap trapping device: 1: glass cooling mantel; 2: vial; 3: glass head connection; 4: nitrogen evacuation pipe; 5: pipe connection to the puff generator; 6: 3-way valve; 7: adsorption tube; 8: glass fibre filter holder; 9: cigarette holder.


Figure 2: Drawing of the custom-designed vial.
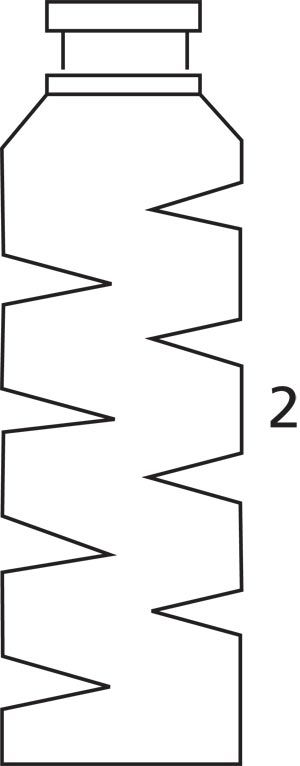
The custom-designed vial had a Vigreux column-like internal geometry to increase flow turbulence and to maximize contact between the vial surface and the aerosol. The external dimensions of the vial corresponded to the model used for headspace analyses with the Turbomatrix 40 Trap (PerkinElmer) headspace sampling device.
To prevent contamination from the laboratory, a glass tube filled with activated charcoal (part 7 in Figure 1) was connected to the switch valve (part 6 in Figure 1). Between each puff, the valve was switched to let pressure equilibrate and avoid any back flush through the glass fibre filter and the cigarette.
At the end of each smoking run, the vial was capped with a Teflon cap. The glass filter contained in the filter holder was transferred into a separate standard headspace vial and capped with a Teflon cap.
This trapping mode enabled the analysis of compounds with a large range of volatility without liquid extraction or dilution. The use of a headspace sampler gave the opportunity to sample the volatile and highly volatile compounds, without extracting the compounds with low volatility.
Headspace Analysis: Headspace sampling was able to capture a fraction of the headspace volume onto a headspace trap prior to thermal desorption and injection onto the chromatographic system via a heated transfer line. The headspace trap was loaded with three different sorbents: Tenax GR (PerkinElmer), Carbotrap and Carboxen (Supelco). The choice of sorbents was made based upon internal (unpublished) and external studies (14), which demonstrated that no universal sorbent was adequate to effectively trap and desorb the compounds of interest belonging to various chemical classes, and that a mixture of sorbents was required.

Figure 3: Technical scheme of a cryo-trap: 1: Metal trap formed by two cylinders separated by isolation material; 2: heating device; 3: electrical connection and type K thermocouple; 4: entrance of the capillary column coming from the valve; 5: exit for N2; 6: exit of the capillary column towards the GC; 7: liquid nitrogen entry. The trap is located on the GC, replacing the classical injectors.
After each smoking run, trapped semivolatile (glass fibre filter in standard headspace vial) and volatile (custom-made headspace vial) fractions were submitted for headspace extraction. The instrument conditions were optimized for each fraction to ensure the effective transfer of trapped smoke constituents. During the release phase, vapour pressure was a key parameter influencing both the diversity and quantity of compounds extracted (15); fine-tuning was required to avoid any segregation of compounds as a result of differing volatilities. The headspace volumes generated from each fraction were trapped concomitantly onto the same headspace trap, and the retained compounds were then released for injection into the chromatographic system by thermal desorption at 210 °C.
The GC–MS system (Perkin Elmer Clarus 500) was modified by the addition of two cryogenic systems (termed cryo-traps), developed and manufactured internally at Philip Morris International for this device (Figure 3), and two electronically actuated valves (4 ports gas valve, 300 psi, 1/16’’, N9302813, PerkinElmer, 8 ports gas valve, 300 psi, 1/16’’, N9302815, PerkinElmer), which were installed in the oven to connect the two different chromatographic columns (1st column: 30 m × 0.32 mm, 1-μm Agilent DB-WAXetr; 2nd column: 30 m × 0.32 mm, 20-μm Agilent HP-PLOT/Q). The two cryogenic systems, cooled using liquid nitrogen, enabled the cryo-focusing of compounds (at a temperature of -80 °C for the one located before the DB-WAXetr column and at -120 °C for the one located before the HP-PLOT/Q column) before introduction to the respective columns. The two switching valves were controlled by the GC–MS events software. These modifications enabled a two-dimensional analysis of cigarette smoke fractions by GC–MS.
The analytical process was initiated by the operator. LabVIEW software (National Instruments) then conveyed this start signal to the GC and to the cryo-traps, and was therefore used as a master controller for the analytical system. The two valves, V1 and V2, were controlled indirectly via the GC programme sequence.
Improved Gas Chromatography, with Dual Columns and Cryogenic Systems

Figure 4: Phase 1.
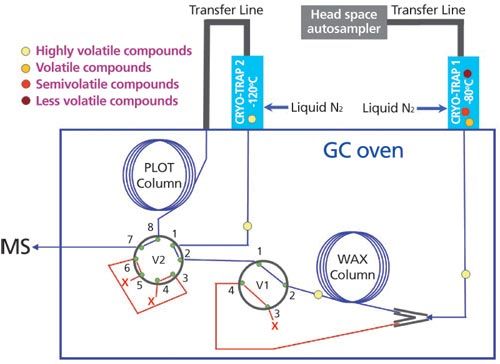
Each analytical run was split in four successive phases to allow adequate separation of compounds in the two cryogenic traps and in the two chromatographic columns.
Phase 1: Sample Introduction and Cryo-Focusing (Figure 4): Following desorption of the headspace trap, which contained headspace constituents from both semivolatile and volatile fraction collections, eluted compounds were focused at -80 °C in the cryo-trap positioned in front of the first column. Highly volatile compounds, which were not retained at this temperature, passed directly through the first chromatographic column. These were trapped by the second cryo-trap cooled at -120 °C, located in front of the second chromatographic column, which was maintained at â120 °C. These two different trapping temperatures led to a split of very volatiles compounds from volatiles, to elute each group of compound selectively on the two dedicated chromatographic columns.
Phase 2: Release from Cryo-Trap #1 and Elution onto the First Column: The first cryo-trap was rapidly heated to 200 °C and a start signal was sent by the software to the GC–MS system to start the chromatographic run. All compounds trapped in cryo-trap #1 were released and introduced onto the WAX column. Analytes with limited affinity for the column eluted rapidly and were trapped in the second cryo-trap. The remaining analytes continued to be separated on the WAX column by their different retention behaviours.
Phase 3:Analysis of Semivolatile Compounds: Valve V2 was switched (by the GC–MS) to isolate cryo-trap #2 (maintained at -120 °C) and the PLOT Q column. This switch connected the exit of the WAXetr column to the mass spectrometer and enabled the analysis of the semivolatile compounds remaining in the chromatographic column. A first heat cycle of a dedicated double heat cycle GC oven temperature programme was used with the following parameters: initial temperature: 41 °C during 5 min, followed by a temperature increase at a rate of 5 °C/min, up to 250 °C.
Phase 4: Release from Cryo-Trap #2 and Analysis of Volatile Compounds: After chromatographic analysis of the semivolatile compounds using the WAX column (30 min run time), the oven was cooled to 60 °C using CO2 to speed the process. For the release of the volatiles, valve V1 was switched to isolate the WAX column and valve V2 was switched back to direct the carrier gas through cryo-trap #2, the PLOT Q column, and into the mass spectrometer. The volatile compounds were then introduced onto the PLOT Q column by rapidly heating cryo-trap #2 to 150 °C and chromatographically separated using the second heat cycle of the GC oven temperature programme with the following parameters: 60 °C during 7.2 min, followed by a temperature increase at a rate of 7 °C/min, up to 250 °C, final temperature maintained during 9 min.
At the end of the analysis, the valve V1 was switched again to allow the carrier gas to pass through both columns and therefore flush the system. This step was done with the oven heated to its maximum temperature, and to allow the valve to be ready for the next run because it was not possible to switch valve V1 after the end of the GC run.
Data acquisition in scan mode was initiated 15 min after the start of each run because no meaningful data were generated during the trapping phase for cryo-trap #2. A second pause in acquisition was also made between 46.8 min and 54 min, during the cooling phase of the oven before the second temperature gradient started.
To compare the described approach with a classical analysis, the cigarette 3R4F (supplied by University of Kentucky) was conditioned according to ISO 3402 standard and smoked using the ISO smoking regime (as described in ISO 3308 standard, 1 puff per min, puff duration: 2 s, puff volume 35 mL, puff profile: bell shape). The particulate phase of the smoke of 10 cigarettes was trapped using a glass fibre filter and the gas phase was bubbled through cooled (-78 °C) ethyl acetate in three small impingers mounted in series. The glass fibre filter was extracted together with the solvent contained in the impingers and was analyzed by GC–MS in scan mode following an internal GC–MS method.
Results and Discussion
A typical total ion chromatogram (TIC) for the classical analysis is presented in Figure 5. One drawback of this technique is linked to the solvent, which prevents the detection of coeluting compounds (volatile compounds). Another issue is compound coelution, which can lead to uncertainty during the peak identification process.

Figure 5: Chromatogram of mainstream smoke from the reference cigarette (3R4F supplied by the University of Kentucky). Ten cigarettes were smoked under ISO smoking regime and trapped on a glass fibre filter followed by three impingers containing each 10 mL ethyl acetate cooled at -78 °C. One microlitre of sample was injected on-column in a DB-FFAP column (30 m à 0.25 mm, 0.25-μm). Analytes: (1) isoprene, (2) solvent, (3) toluene, (4) limonene, (5) nicotine, (6) neophytadiene, (7) triacetin, and (8) glycerin.
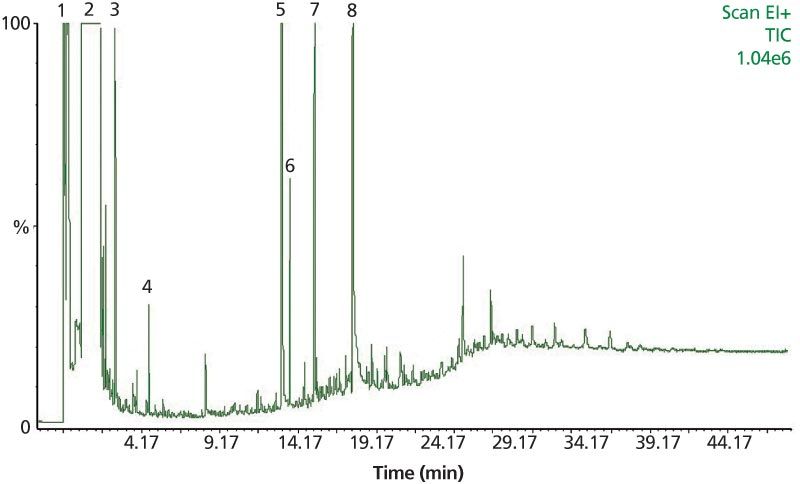
Figure 5: Chromatogram of mainstream smoke from the reference cigarette (3R4F supplied by the University of Kentucky). Ten cigarettes were smoked under ISO smoking regime and trapped on a glass fibre filter followed by three impingers containing each 10 mL ethyl acetate cooled at -78 °C. One microlitre of sample was injected on-column in a DB-FFAP column (30 m × 0.25 mm, 0.25-μm). Analytes: (1) isoprene, (2) solvent, (3) toluene, (4) limonene, (5) nicotine, (6) neophytadiene, (7) triacetin, and (8) glycerin.
The innovative aspect of the new platform lies in the use of a dedicated column selected specifically for the volatility of each group of compounds. Using this setâup, the chromatogram showed first the semivolatile compounds eluted on the 30 m × 0.32 mm, 1-μm column (Figure 6) and later the volatile ones eluted on the 30 m × 0.32 mm, 20-μm column (Figure 7). The split between semivolatile and volatile compounds was handled using two specific chromatographic columns with dedicated oven programmes, allowing an improvement in the chromatography.

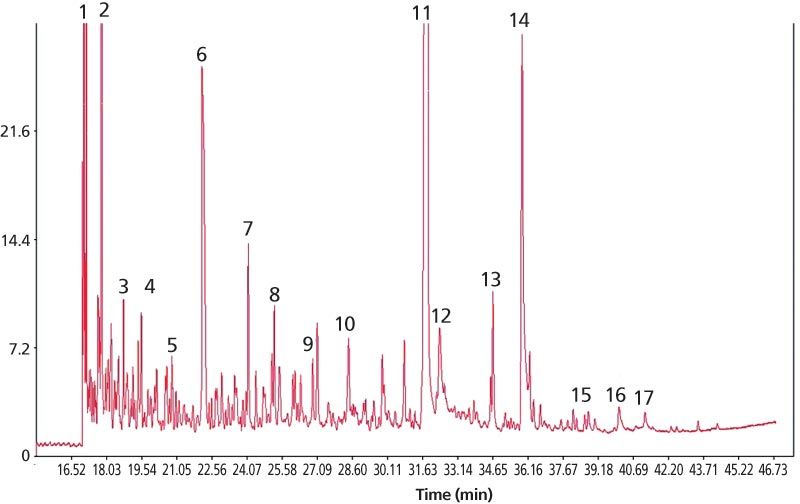
Figure 6: Chromatogram of the semivolatile part of the mainstream smoke from one single reference cigarette (3R4F, supplied by the University of Kentucky) after separation on a DB-WAXetr column. Analytes: (1) ethylbenzene, (2) limonene, (3) styrene, (4) acetol, (5) 2-cyclopentene-1-one, (6) acetic acid, (7) Propanoic acid, (8) propylene glycol, (9) 2-furanmethanol, (10) solanone, (11) nicotine, (12) neophytadiene, (13) phenol, (14) triacetin, (15) myosmine, (16) glycerin, and (17) nicotyrine.
A clear improvement in selectivity was apparent when using the cold trap coupled with the headspace sampling compared with the traditional analysis. Peaks are properly resolved and can be identified with improved certainty. In addition, the second part of the chromatogram (starting at approximately 53 min) allowed the identification of numerous volatile compounds that are masked by the solvent peak when using conventional GC–MS analysis. For example, Figure 7 demonstrates that compounds such as chloromethane, HCN, acetaldehyde, ethanol, acetonitrile, propionaldehyde, and acetone were clearly separated.

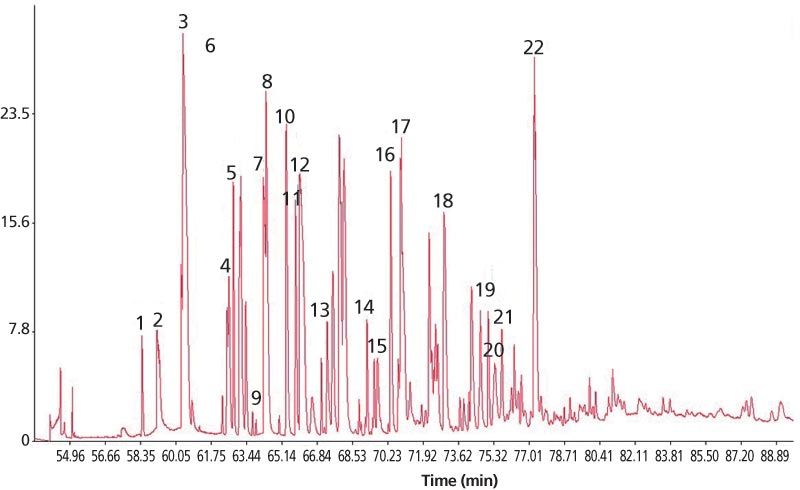
Figure 7: Chromatogram of the volatile part of the mainstream smoke from one single reference cigarette (3R4F, supplied by the University of Kentucky) after separation on the HP-PLOT/Q column. Analytes: (1) chloromethan, (2) HCN, (3) acetaldehyde, (4) 2-methyl-1-propene, (5) 1-3, butadiene, (6) 2- butene, (7) ethanol, (8) acetonitrile, (9) 2- butyne, (10) furan, (11) propionaldehyde, (12) acetone, (13) acrylonitrile, (14) propanenitrile, (15) metacrolein, (16) methylfuran, (17) mix of butyraldehyde and methyl ethyl ketone, (18) benzene, (19) 2-pentanone, (20) 2,3-pentanedione, (21) dimethyl disulphide, and (22) toluene.
Furthermore, the sensitivity of the technique allows the analysis of cigarette mainstream smoke from a single cigarette, therefore reducing the number of cigarettes required, while increasing the level of information gained.
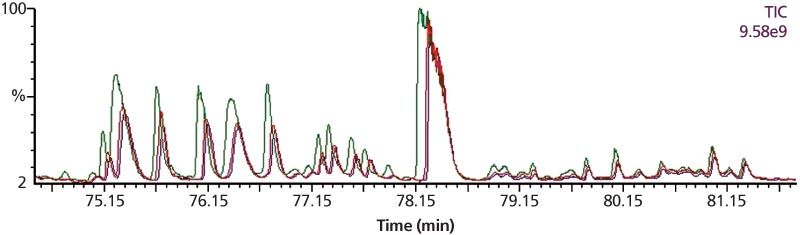
The automation of the analytical process ensured reproducible retention times across several different runs (Figure 8) and displayed very volatile compounds on the 30 m × 0.32 mm, 20-μm column. The same reproducibility in retention times was observed on the 30 m × 0.32 mm, 1-μm column.

Figure 8: Overlay of the volatile part of three chromatograms from reference cigarettes (3R4F, supplied by the University of Kentucky).
Conclusion
The platform for trapping and analysis allowed the characterization of cigarette smoke in an efficient and simple way. The trapping device is an improvement compared to classical liquid trapping because of the absence of solvent, which reduces sample preparation, prevents any chemical interaction between solvent and compounds of interest, and allows determination of chemical species which are usually masked by the solvent peak. In addition, no dilution is performed during sample preparation; therefore, the analysis can be performed on a single cigarette. Improved sensitivity and resolution enables a better and more comprehensive characterization of cigarette mainstream smoke.
The method was focused on qualitative aspects to facilitate compound identification, rather than to quantify them. Because compounds may have very different vapour pressures under the chosen conditions, headspace sampling would not be suitable for direct quantification of samples.
The improved chromatographic analysis allowed an easier identification of compounds because of the improved peak capacity provided by the hyphenation of the two columns, which led to a better peak separation. Retention times were reproducible because the complete process was fully automated. This reproducibility also allows the creation of custom libraries to improve identification certainty.
References
(1). R.R. Baker, in Tobacco: production, chemistry and technology, D.L. Davis and M.T. Nielsen, Eds. (Blackwell Science Ltd, Oxford, 1999), p. 398–439.
(2) M.F. Borgerding, J.A. Bodnar, H.L. Chung, et al., Food and Chemical Toxicology36, 169–182 (1997).
(3) T. Perfetti and A. Rodgman, Beiträge zur Tabakforschung International24(5), (May 2011).
(4) International Organization for Standardization (ISO): ISO 4387:2000, Cigarettes -- Determination of total and nicotine-free dry particulate matter using a routine analytical smoking machine. Geneva, Switzerland.
(5) International Organization for Standardization (ISO): ISO 22634:2008, Cigarettes -- Determination of benzo[a]pyrene in cigarette mainstream smoke -- Method using gas chromatography–mass spectrometry. Geneva, Switzerland.
(6) Ji-Zhou Dong, J. Neil Glass, and Serban C. Moldoveanu, Journal of Microcolumn Separations 12(3), 142–152 (2000).
(7) J.Z. Dong and S.M. Debusk, Chromatographia71(3/4), (2010).
(8) N. Mottier, F. Jeanneret, and M. Rotach, Journal of AOAC International 93(3), (2010).
(9) Health Canada: Determination of Ammonia in Mainstream Tobacco Smoke, Official Method T-101 (1999) http://laws-lois.justice.gc.ca/eng/regulations/SOR-2000-273/page-14.html
(10) Health Canada: Determination of Selected Carbonyls in Mainstream Tobacco Smoke, Official Method T-104 (1999) http://laws-lois.justice.gc.ca/eng/regulations/SOR-2000-273/page-14.html
(11) N. Plata, I. Hofer, S. Roudier, and J.P. Schaller, Journal of Aerosol Science37(12), 1871–1875, (2006).
(12) International Organization for Standardization (ISO): ISO 3402:1999, Tobacco and tobacco products -- Atmosphere for conditioning and testing. Geneva, Switzerland.
(13) International Organization for Standardization (ISO): ISO 3308:2000, Routine Analytical Smoking Machine -- Definition and Standard Conditions. Geneva, Switzerland.
(14) M. Schneider and K.-U. Goss, Analytical Chemistry81, 3017–3021 (2009).
(15) B. Kolb and L.S. Ettre, Static Headspace-Gas Chromatography: Theory And Practice (Wiley, UK, 2nd ed., 2006).
Jean-René Crudo is an associate scientist at Philip Morris International (Research and Development, Neuchâtel, Switzerland). His analytical focus is on the development and application of GC–MS methods focusing on the qualitative and quantitative analysis of complex matrices.
Emmanuel Rouget is a scientist at Philip Morris International (Research and Development, Neuchâtel, Switzerland). His main research interests are related to the development and application of GC–MS techniques concerning qualitative and quantitative analysis of molecules.
Michel Rotach is a senior scientist at Philip Morris International (Research and Development, Neuchâtel, Switzerland). His main activities involve providing scientific advice to sustain the development of Reduced Risk Product (RRPs), perform data analysis, and provide scientific guidance.


















Study Examines Impact of Zwitterionic Liquid Structures on Volatile Carboxylic Acid Separation in GC
March 28th 2025Iowa State University researchers evaluated imidazolium-based ZILs with sulfonate and triflimide anions to understand the influence of ZILs’ chemical structures on polar analyte separation.

