A Hydrophilic Interaction Chromatography Method for the Purity Analysis of Cytosine
LCGC Europe
Cytosine (chemical name 4-amino-2-hydroxypyrimidine) is a pyrimidine derivative with a hetereocyclic aromatic ring and two substituents (amine and keto groups) attached and is a polar compound of significant biological and pharmaceutical interest. In response to the intended use of bulk cytosine as a raw material in pharmaceutical manufacturing, a method for the determination of the purity of cytosine was developed.
Cytosine (chemical name 4-amino-2-hydroxypyrimidine) is a pyrimidine derivative with a hetereocyclic aromatic ring and two substituents (amine and keto groups) attached and is a polar compound of significant biological and pharmaceutical interest. In response to the intended use of bulk cytosine as a raw material in pharmaceutical manufacturing, a method for the determination of the purity of cytosine was developed.
Chromatographic separations of nucleobases, including cytosine, and their derivatives have traditionally been performed through the use of ion-exchange chromatography,1,2 specifically for preparative applications. Analytical techniques that have been used for the separation of nucleobases have included gas chromatography (GC).3,4 and micellar electrokinetic capillary chromatography (MECK).5 Disadvantages of these approaches include the need for sample derivatization and the need for custom-built instrumentation, respectively. High-resolution liquid chromatography (LC) analyses of cytosine have been difficult because the compound is highly polar and cannot be analysed effectively by conventional reversed-phase (RP) chromatography. The use of ion-pair reagents has been demonstrated to provide acceptable retention and separation of this compound.6 However, experience with the mobile phases containing octanesulphonic acid revealed the presence of a significant number of artifacts in the chromatogram, most likely caused by impurities in the ion-pair reagent.7 Additionally, the capability of the method for interface with electrospray ionization-mass spectrometry (ESI-MS) detection for the characterization of related impurities was desired. ESI-MS is not feasible with nonvolatile ion-pair agents such as octanesulphonic acid and not optimal with volatile ion-pair agents, such as trifluoroacetic acid, which cause ESI signal suppression.8
To meet these needs, a hydrophilic interaction chromatography (HILIC) method was developed and interfaced with ESI-MS. HILIC is a technique that has been demonstrated to provide retention and resolution of highly polar compounds.9 HILIC provides a retention mechanism somewhat similar to traditional normal-phase (NP) chromatography, but offers the benefit of compatibility with water-soluble analytes through the inclusion of significant aqueous portions in the mobile phase. The technique has been applied to determine polar pharmaceuticals and their impurities10,11 and also for active pharmaceutical ingredient starting materials.12 The chromatography of cytosine and its aqueous degradation product, uracil, have been previously evaluated by HILIC10,13 and in this paper, we present a purity method for HILIC validated according to the ICH guidelines, including a comprehensive evaluation of robustness of the method in response to small changes in column temperature, mobile phase pH, mobile phase buffer concentration, injection volume and flow-rate. HILIC also has been interfaced successfully with ESI-MS for on-line characterization and quantification.14–19 In this study, the cytosine purity method was also interfaced successfully with ESI-MS to help confirm peak identity and to enable characterization of unknown contaminants.
Experimental
Materials: Acetonitrile was purchased from Burdick and Jackson (Muskegon, Michigan, USA). Water was purified using an Elga PureLab Ultra water purification system (Vivendi Water Systems, Buckinghamshire, UK). Formic acid (99%) was purchased from Acros Organics (Morris Plains, New Jersey, USA) and sodium hydroxide and ammonium hydroxide were purchased from Red Bird Chemicals (Osgood, Indiana, USA). Cytosine and uracil were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, Wisconsin, USA) and 7-amino-1,2-dihydro-2-oxo-pyridol[2,3-d]pyrimidine (7-ADOP) was provided by Eli Lilly and Company (Indianapolis, Indiana, USA).
Equipment: Separations were performed using a 250 mm × 4.6 mm, 5 μm dp TSKgel Amide-80 column from Tosoh BioSciences (Montgomeryville, Pennsylvania, USA), a 250 mm × 4.6 mm, 5 μm dp Spherisorb Amino column from Waters Corporation (Milford, Massachusetts, USA) and a 250 mm × 4.6 mm, 5 μm dp MacMod Zorbax Amino column from MacMod Analytical (Chadds Ford, Pennsylvania, USA) on an Agilent (Wilmington, Delaware, USA) 1100 HPLC system, with a binary pump and fixed-wavelength detection at 260 nm. Data acquisition and processing were conducted using Waters Millennium software. For MS analysis, a Waters 2695 Separations Module, a model 2996 diode-array detector and an EMD 1000 mass spectrometer scanning from 75–1000 m/z with scan speed of 2 scans/s, capillary and cone voltage potentials of 3500 and 40 V, respectively, with positive–negative ion switching were employed.
Methods: Mobile phase A was prepared by adding 2.0 mL of formic acid per litre of purified water and adjusting the pH to 3.5 with 5 N sodium hydroxide. For HILIC–ESI-MS analyses, ammonium hydroxide was substituted for sodium hydroxide. Mobile phase B was acetonitrile. Sample diluent was prepared by mixing mobile phases A and B in a 1:4 ratio. The system suitability mixture was prepared in sample diluent at a concentration of 0.5 mg/mL cytosine and 0.01 mg/mL of uracil and 7-ADOP, with sonication. Sample solutions were prepared at a concentration of 0.5 mg/mL in sample diluent and were spiked with 7-ADOP at levels of 0.02%, 1.07% and 4.16% (w/w) for validation experiments.
Samples held at ambient temperature in the autosampler were injected at a 10 μL volume. The column was incubated at 30 °C and the flow-rate was 1.0 mL/min. The gradient was as follows: The initial condition was 5% mobile phase A. After injection, initial conditions were held for 5 min, increased to 25% mobile phase A at 2%/min over 10 min, held for 5 min and then returned to the starting conditions for a 10 min reequilibration at starting conditions. The total run time was 35 min.
For HILIC–ESI-MS analyses, the effluent stream was split postcolumn to the MS system resulting in a 300 μL/min flow into the ESI interface.
Results and Discussion
HILIC chromatography was employed through the use of the three stationary phases to assess selectivity using a mixture of cytosine and two critical related impurities, 7-ADOP and uracil. 7-ADOP is the primary impurity observed in commercially available cytosine. Cytosine is inherently unstable and can convert into uracil via spontaneous deamination. Therefore, uracil is considered the primary degradation product of cytosine.
The results of the evaluation of separation selectivity are displayed in Figure 1. In this experiment, the gradient conditions described in the Experimental section (5 min hold at 5% aqueous, 5–25% aqueous gradient in 10 min, followed by a 5 min hold at 25%) were employed with each column listed in the Experimental section. In each instance, the retention of uracil, 7-ADOP and cytosine were centred acceptably within the retention window, indicating the robustness of the method in response to column packing changes. Using the Tosoh Amide 80 column, the following experiments were performed to validate application of this method for quantitative determination of cytosine purity.



Figure 1
The linearity of the cytosine main peak was demonstrated from 50% to 125% of the nominal concentration of 0.5 mg/mL with a correlation of 0.999. The y-intercept was approximately 1.0% of the value at the nominal concentration. The linearity of 7-ADOP and uracil were demonstrated from approximately 0.1 μg/mL to 20 mg/mL [0.02–4.0% (w/w) of cytosine at nominal concentration]. Both of these correlation coefficients were 0.9999, and the y-intercepts were less than 0.2% of the nominal level of 1.0% (w/w) cytosine.


Table 1: Experimental design of the robustness study. ,N. refers to the default parameter setting as indicated in the method.
For triplicate preparations at each level, 7-ADOP mean recoveries of 98.4%, 100.5% and 99.9% were determined when samples in the absence and presence of matrix (cytosine at the nominal concentration) were spiked with 0.02%, 1.1% and 4.2% (w/w) of 7-ADOP to cytosine, respectively.


Table 2: Experimental design setup for the robustness study.
For determination of selectivity, baseline resolution was established for cytosine, 7-ADOP and uracil, as demonstrated in Figure 1. No significant background interferences from the sample diluent (as determined through an injection of the sample solvent blank) or spiked samples were observed.


Table 3: Results of the robustness study.
For an evaluation of robustness, an experimental design focusing on estimation of factor main effects was employed. Nine method set-ups were employed to determine the effects of variations in the parameters of flow-rate, column temperature, injection volume, mobile phase pH and mobile phase buffer concentration upon the chromatography. See Tables 1 and 2 for a description of the experimental design and Table 3 for a summary of the results. To isolate the contributions of each of the individual factors, the average performance of cytosine retention time, cytosine tailing factor and 7-ADOP:cytosine peak resolution were compared at high and low values of each parameter (see Tables 4–6). As expected, the largest effect observed in this experiment was that of flow-rate upon cytosine retention time. The method validation robustness study confirmed that the method was robust when operated within the range of method factors.


Table 4: Effects of factors upon cytosine retention time.
For determination of method precision, six replicate cytosine samples spiked with 0.25%, 0.50% and 1.0% impurities (as previously used for the determination of accuracy) were also assessed for repeatability. Mean values for total related impurities in those samples were 0.30%, 0.57% and 1.09%, respectively. The relative standard deviation (RSD) of each result was 0.4%, 0.1% and 0.7%, respectively. Six replicates measured for largest individual impurity (in this instance the 7-ADOP) yielded a mean value of 0.24%, with a RSD of 1.1%. The 95% upper confidence interval of the RSD was 2.3% for the largest individual impurity.


Table 5: Effects of factors upon cytosine peak tailing.
Intermediate precision was assessed through the use of cytosine solutions spiked with 7-ADOP to a level of approximately 0.50% (w/w). These solutions were analysed by two analysts over two days, each using two different chromatography system set-ups. Total related impurities results averaged 0.53%, with a RSD of 2.7%. Largest individual impurity results averaged 0.33%, with a RSD of 2.9%. No other related impurities were present at or above 0.25%. The 95% upper confidence interval of the RSD was 4.3% for the largest individual impurity and 4.0% for total related impurity.


Table 6: Effects of factors upon 7-ADOP:cytosine resolution.
To further demonstrate the reproducibility of the HILIC analyses, cytosine peak area and retention time were determined run-to-run across a series of 100 replicate injections, as displayed in Figure 2. For the entire data set, values of %RSD for peak area and retention time were calculated to equal 0.73 and 0.12, respectively.

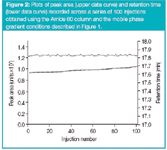
Figure 2
Cytosine samples prepared according to the method were found to be stable for up to 30 days when stored at ambient temperature or refrigerated conditions (4 °C). System suitability solutions also were stable for up to 30 days when stored at room temperature. Precipitation of the 7-ADOP at 4 °C indicated that storage of the system suitability solution at refrigerated conditions was not appropriate.
The evaluation of a HILIC–ESI-MS method was achieved through substitution of sodium hydroxide with ammonium hydroxide in the mobile phase to provide a completely volatile mobile phase. Successful performance of this system was confirmed through evaluation of the standards of the individual compounds and it is important to note that the minor change in mobile phase constituent had no discernable impact upon the chromatography. An overlay of the positive ion total ion chromatograms obtained for the standards is displayed in Figure 3, along with the structures of the individual analytes. Through the use of ESI-MS, the identities of each component were confirmed through their corresponding positive ion mass spectra (See Figure 3 insets). In each spectrum, the highest intensity ion corresponded to the protonated molecular ion.


Figure 3
Conclusions
A HILIC method was developed successfully and validated for the determination of cytosine purity. The method was validated for precision, accuracy, robustness, selectivity, linearity and stability, as per ICH guidelines for chromatographic methods. The method demonstrated acceptable robustness, indicating that HILIC can be considered an appropriate tool for use within a rigorous environment such as that present within a quality control laboratory. The method was interfaced to ESI-MS detection and was used successfully to confirm the identity of selected related impurities.
Mark Strege, Charles Durant, John Boettinger and Michael Fogarty, Lilly Research Laboratories A Division of Eli Lilly and Company Lilly Corporate Center, Indianapolis, Indiana, USA
References
1. H. Yuki et al., Chem. Pharm. Bull. , 25(11), 2827–2830 (1977).
2. P. Linssen et al., J. Chromatogr. , 232(2), 424–429 (1982).
3. K. Schram, Y. Taniquchi and J. McCloskey, J. Chromatogr. A , 155(2), 355–361 (1978).
4. C. Gehrke and A. Patel, J. Chromatogr. A, 123(2), 335–345 (1976).
5. G. Chen, X. Han and L. Zhang, J. Chromatogr. A , 954(1–2), 267–276 (2002).
6. R. Schilsky and F. Ordway, J. Chromatogr. , 337(1), 63–71 (1985).
7. M. Fogarty, unpublished results.
8. S. Gustavsson et al., J. Chromatogr. A, 937(1–2), 41–47 (2001).
9. A. Alpert, J. Chromatogr. , 499, 177–196 (1990).
10. B. Olsen, J. Chromatogr. A, 913, 113-122 (2001).
11. X. Wang, W. Li and H. Rasmussen, J. Chromatogr. A , 1083, 58–62 (2005).
12. P. Gavin et al., J. Pharm. Biomed. Anal. , 41, 1251–1259 (2006).
13. S. Bajad et al., J. Chromatogr. A , 1125, 76–88 (2006).
14. M. Strege, Anal. Chem. , 70, 2439–2445 (1998).
15. M. Strege, Am. Pharm. Rev. , 2, 53–58 (1999).
16. H. Koh, A. Lau and E. Chan, Rapid Commun. Mass Spectrom. , 19, 1237–1244 (2005).
17. Y. Hsieh and J. Chen, Rapid Commun. Mass Spectrom. , 19, 3031–3036 (2005).
18. P. Uutela et al., Rapid Commun. Mass Spectrom. , 19, 2950–2956 (2005).
19. J. Bengtsson, B. Jansson and M. Hammarlund-Udenaes, Rapid Commun. Mass Spectrom., 19, 2116–2122 (2005).














New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.






