Optimization Approaches in the Routine Analysis of Monoclonal Anitbodies by Capillary Electrophoresis
LCGC Europe
Electrophoretic techniques, such as sodium dodecyl sulphate polyacrylamide (SDS-PAGE) and isoelectric focusing (IEF) gels, have traditionally been part of release testing in the biopharmaceutical industry. However, these slab gel methods are inconvenient and irreproducible because of the staining/destaining steps used in analyte detection, the use of toxic reagents and high intra- and inter-gel effective mobility variability.
Electrophoretic techniques, such as sodium dodecyl sulphate polyacrylamide (SDS-PAGE) and isoelectric focusing (IEF) gels, have traditionally been part of release testing in the biopharmaceutical industry. However, these slab gel methods are inconvenient and irreproducible because of the staining/destaining steps used in analyte detection, the use of toxic reagents and high intra- and inter-gel effective mobility variability.
As a result, capillary electrophoresis (CE) methods such as capillary electrophoresis sodium dodecyl sulphate (CE–SDS), capillary isoelectric focusing (CIEF) and capillary zone electrophoresis (CZE) have emerged as practical replacements of the slab gel format for many applications in biotechnology
In addition, with the advances made in reagents and commercially available instrumentation, CE has expanded its important role in biopharmaceutical analysis by providing complementary information of the physicochemical attributes of therapeutic proteins in terms of charge, hydrophobicity and size.
Furthermore, with respect to quality control (QC) laboratories, CE has become an integral part of the specifications of therapeutic proteins. The QC specifications consist of a selected list of methods with relevant acceptance criteria, ensuring the safety, identity, purity and composition of a drug substance and drug product.
This "CE Currents" describes the advances in three CE applications for the analysis of protein-based pharmaceuticals, and their role in the overall QC specifications at Genentech Inc. The first application illustrates the optimization of a generic and quantitative CE–SDS assay with LIF detection with improved accuracy and precision to assess the size heterogeneity of recombinant monoclonal antibodies (rMAbs). The second section focuses on the use of imaged CIEF to determine charge heterogeneity of rMAbs. The third section focuses on the practical applications of interfacing CE to mass spectrometry (MS) for the profiling of rMAb glycosylation. Finally, we summarize in the last section additional considerations to be taken with respect to CE training and instrument maintenance to ensure high success rates while applying these practical and complex methods in a routine QC laboratory.
Optimization of an rMAb Sample Preparation Scheme for Quantitative CE–SDS Analysis with LIF Detection
CE–SDS has been recognized as a robust technique to determine and/or confirm the apparent molecular weight of proteins and to evaluate the size heterogeneity and purity of biologics.1–4 The successful performance of a commercial rMAb CE–SDS based method was recently reported when used by eleven independent biopharmaceutical companies and a regulatory authority.5
The rMAb standard employed contained a mixture of light chain (LC), non-glycosylated heavy chain (NGHC) and heavy chain (HC) components. The high separation power of CE–SDS was demonstrated by showing baseline resolution between HC and NGHC species present at a low level (less than 5%). The relative standard deviation (RSD%) for the relative migration time of those components was ~2%. Moreover, the percentage of peak area quantification of the rMAb sample components was comparable across all organizations with RSD% values of less than 9%.5
It is important to indicate that an advantage of CE–SDS over other size-based separation methods such as high-performance size-exclusion chromatography (HPSEC) and SDS–PAGE is the improved resolution of more closely related size-variants of proteins. Therefore, the CE–SDS analysis of reduced rMAb samples gives more reliable, accurate and reproducible quantification of the glycolysation site occupancy. Because of these features, CE–SDS is also expected to be a routine technique for the quantitative analysis of the size heterogeneity or fragmentation of non-reduced rMAb samples.
However, traditional sample preparation conditions to form SDS–protein complexes prior to electrophoretic analysis includes heat treatment at elevated temperatures (e.g., 90°C). In the case of non-reduced rMAbs, this could lead to sample preparation artifacts in the form of thermally induced fragmentation attributed to disulphide reduction and exchange reactions.1,4 It was also reported that high pH conditions during heat treatment also enhanced the fragmentation of SDS-rMAb complexes.3 These artifacts significantly alter the true representation of the size heterogeneity of a protein and also increase the variability of quantitative CE–SDS methodologies of non-reduced samples. As a result, Genentech used non-reduced CE–SDS analysis as a qualitative assay to demonstrate that the molecular mass distribution of a bulk manufacture run was comparable to that of a well-characterized reference material, similar to the practice used in SDS–PAGE. Moreover, to improve the sensitivity of UV absorbance, a common detector in CE–SDS, we demonstrated that precolumn labelling of recombinant protein products with 5-carboxytetramethylrhodamine succinimidyl ester (5-TAMRA.SE) and LIF detection can be used for CE–SDS analyses as a replacement for silver-stained SDS–PAGE to detect low-level impurities and rMAb size-variants.1 The analysis of trace-level rMAb variants and process impurities at levels as low as 50 ppm were reported using CE–SDS with LIF detection.
To greatly decrease any thermally induced fragmentation of non-reduced unlabelled and labelled rMAbs, the sample preparation conditions before CE–SDS analysis were systematically optimized.4 The inclusion of an alkylation step with 40 mM iodoacetamide (IAM) during heat treatment of the non-reduced rMAb sample and optimization of the incubation temperature and time to 70 °C for 5 min, in the presence of SDS, significantly suppressed the extent of thermally-induced fragmentation. The quantitative studies of our work also demonstrated that CE–SDS sample buffers at pH levels of 6.5 further decreased the induced fragmentation of non-reduced rMAb samples and gave improved sample stability.4 As shown in Figure 1 (top trace), a significant decrease of the peak areas corresponding to the rMAb fragments was observed for a sample treated with the optimized sample preparation scheme when compared with an rMAb control sample. The control sample was prepared using traditional sample preparation conditions that included using 100 mM Tris-HCl buffer pH 9.0 as CE–SDS sample buffer and incubating the sample at 90°C for 5 min. The corrected percentage peak area (%CPA) of the intact antibody increased from 90.0% in the control sample to 98.5 % in the sample containing 40 mM IAM, reconstituted in 86 mM citrate–phophate pH 6.5 and incubated at 70°C for 5 min. The final SDS concentration in all the samples was 1% (w/w). Similar results were obtained when iodoacetic acid (IAA) was used as the alkylating agent. However, IAM was the reagent of choice because solutions of IAA were less stable as indicated by the appearance of a yellow colouration after a few minutes.



Figure 1
The addition of the alkylating agent to reduced labelled-rMAb samples was not required. As depicted in Figure 1 (bottom trace), upon the addition of 1M dithiothreitol (DTT) the rMAb is reduced to light chain (LC), non-glycosylated heavy chain (NGHC), heavy chain (HC) and other small antibody fragments. The CE–SDS profiles of the reduced samples prepared with 40 mM IAM or IAA in the 85 mM citrate-phosphate pH 6.5 were comparable with the control sample prepared as indicated above. The results of the precision studies of the optimized CE–SDS method are shown in Table 1. For repeatability, six individual sample preparations were analysed under both non-reducing and reducing conditions on one day with one injection per sample.
As shown in Table 1, the RSD value of the corrected percentage peak area for the intact antibody of non-reduced samples was only 0.12%. The RSD value was &λt; 0.85% for the corrected percent peak area of the LC and HC in the reduced samples. The intermediate precision study was performed by two analysts in different laboratories. They assayed the samples under both non-reducing and reducing conditions on three separate days using separate instruments, reagents and capillaries. The RSD values were 0.45% for the corrected percent peak area of the intact antibody, 2.39% and 1.42% for the LC and HC, respectively. Taken together, the data demonstrate that CE–SDS with LIF detection is a reproducible and robust technique to assess the size heterogeneity of rMAbs. This methodology is routinely used at GNE for the characterization and lot release of therapeutic rMAbs.

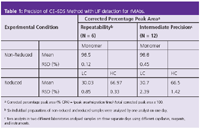
Table 1
Imaged CIEF
Regulatory agencies require the assessment of the charge heterogeneity of biotechnology products. Charged variants of recombinant protein products often include post-translational modifications such as sialylation, phosphorylation, deamidation and C-terminal lysine residues. Such variants are often monitored by the widely used ion-exchange chromatography (IEC) techniques. However, the development of an IEC method is typically extensive and product-specific. IEC assays require the optimization of complex separation parameters including column, mobile phase composition, pH, salt, temperature and gradient. CE techniques, such as CZE and CIEF, offer the advantages of developing generic methods for multiple products and faster analysis time, which is desirable in a high-throughput process development laboratory.
In CZE, analytes are separated from each other based on the differences in their electrophoretic mobilities; therefore, this technique also introduces a size-based element to the separation. Whereas, the ability of CIEF to separate solely based on charge may have an added advantage in isolating charge variants of rMAbs.
CIEF is usually performed on commercial CE instruments, which have a 20–60 cm long capillary and on-column ultraviolet UV absorbance detector. After focusing, the focused protein zones are moved through the detector point by chemical, electrophoretic or hydrodynamic mobilization. In this "two-step" CIEF mode, problems associated with the mobilization process may be encountered, including long analysis time, distortion of pH gradient and uneven resolution because of a non-uniform mobilization speed.
The requirement of the mobilization step can be eliminated using whole-column imaging detection via charged coupled device (CCD) camera. This technique is called imaged CIEF, or iCIEF, and has been recently commercialized by Convergent Biosciences.6 This technique could have the added advantages of robustness because of absence of moving parts typical of conventional CIEF instrumentation, reduced analysis time (less than 15 min per sample), and allowing the user to monitor the focusing process in real-time leading to faster method development. Although iCIEF is still a relatively new technique, the advantages of simplicity and fast analysis time make it more suitable for QC environments and the high-throughput needs in process development. The potential of iCIEF to assess the overall rMAb charge distribution is demonstrated in Figure 2. The electropherogram shows the various isoforms of the antibody between the isoelectric points (pI) markers 7.6 and 9.7. The pI values of the rMAb peaks, shown in the x-axis, were calculated using two-point calibration with the pl markers 7.6 and 9.7. The main peak or 0-Lys form of the rMAb appears at pI 9.2.

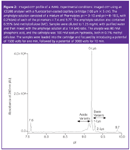
Figure 2
The more basic species with one and two C-terminal Lys residues at the heavy chain are baseline-resolved from each other and the main peak. In addition, the acidic variants were also observed before the 0-Lys peak. Table 2 presents a summary of the reproducibility studies of the iCIEF method to assess the charge variants of the rMAb in Figure 2. The low %RSD values for all the peaks demonstrate that iCIEF can be used in QC as a technique to monitor the charge heterogeneity of protein therapeutics.

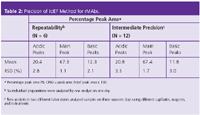
Table 2
On-Line CE–MS for Direct Characterization of N-Linked Glycans
Of the many post-translational modifications present on therapeutic proteins, glycosylation has received considerable attention in the field of proteomics. In the biotechnology industry, it is well known that carbohydrates can play a significant role in the activity and efficacy of a glycoprotein and must be carefully monitored.7–9
Even minor changes in glycan distribution (such as increased afucosylation) can have effects on the activity of a biopharmaceutical product.10–12 Therefore, advances are continually being made in the biopharmaceutical industry to reduce glycan heterogeneity and improve analytical assays used to identify and quantify minor changes across products and/or batches.13
However, the task of glycoprotein characterization remains an analytical challenge because of the immense heterogeneity of these species. Genentech has been using CE-based methods for profiling glycosylation for several years because of the advantages of automation, short analysis times and the ability to separate and quantify isomeric species. The coupling of CE with on-column laser-induced fluorescence (LIF) detection also has the advantage of highly sensitive detection. The primary assay used for profiling rMAb glycosylation involves releasing N-linked carbohydrates with PNGase-F, labelling with the fluorophore 8-aminopyrene-1,3,6-trisulphonate (APTS) and analysis by CE-LIF. APTS was chosen because of the low-picomole sensitivity attainable for APTS- derivatized carbohydrates.14 An earlier version of the assay described by Ma and Nashabeh,15 in which CE-LIF was used for profiling and quantification of rituximab glycans including major species G0, G1, G1'and G2. Currently, a new version of this assay has been validated and is used routinely in a high-throughput format for product characterization, clone selection and lot release of recombinant monoclonal antibody (rMAb) therapeutics.
However, one disadvantage of CE-based methods is the lack of routine detection methods that can provide direct structural information on migrating species. For our glycan assay, characterization was primarily performed using one of two methods:
1. Co-migration with commercially available standards.
2. Exoglycosidase treatment, followed by co-elution with standards or examination of migration shift.
These methods can be particularly labour-intensive and do not provide any direct information on glycan structure. As a result of the more routine use of this assay in all stages of product development, we have been exploring new methods that can be used to characterize new or existing peaks that may be present.
Coupling CE to on-line mass spectrometry (MS) presents an attractive option, allowing direct mass information to be obtained on migrating components.16 Direct CE–MS coupling is usually performed via elecrospray ionization (ESI) and requires an interface that can provide high sensitivity without degrading the separation quality. Peak broadening in CE–MS have been reported as a result of parabolic flow from pressure differences between the capillary ends, dead volume associated with the CE–MS interface and the lack of temperature control at the detector end.17,18 As a result, routine applications of CE–MS are relatively few in the biopharmaceutical industry and most reports have shown little emphasis in evaluating this technique as a robust and reproducible analytical technique to establish the identity of the peaks in the CE–UV or CE–LIF electropherograms. Our group performed numerous studies aimed at optimizing this technology towards routine use in process development and presented several relevant applications in the analysis of peptides, glycopeptides and carbohydrates.19 The current research in our group expands the development of a robust CE–MS methodology for the characterization of the glycan peaks observed in the CE–LIF assay. For MS interfacing, the end of the CE capillary is installed into a CE–MS sprayer (Agilent Technologies, Wilmington, Delaware, USA), allowing for a sheath liquid to be added coaxially. Mass spectrometry detection is achieved using a Bruker MicrOTOF accurate mass MS instrument (Bruker Daltonics, Germany), equipped with an orthogonal, at-ground electrospray ionization (ESI) source.
It is important to mention that using an MS instrument with the spray needle grounded allows the use of high CE currents without the risk of instrument damage. As a result, concentrated buffer systems can be used leading to high-efficiency separations. In addition, the reproducibility and robustness of the CE–MS system is significantly improved.
The ultimate goal is to achieve a similar CE separation profile with mass spectrometric detection as that obtained by the original LIF method to facilitate direct peak identification. To accomplish this, the CE–LIF method was modified to allow MS coupling while ensuring that any differences between the CE–LIF and CE–MS profiles could be attributed only to minor changes in resolution.
A typical CE–LIF glycan profile for an rMAb is shown in Figure 3 (top trace). In this profile, major glycan species G0, G1, G1'and G2 can be easily resolved, as well as several unidentified minor peaks at ;9.7 min. Because of the nature of MS detection, there are several assay parameters that must be modified to use this technique. These modifications include (1) removing the excess salts/label from APTS-labelled glycan samples, which interfere with MS detection and (2) increasing the length of the CE capillary from 60 cm total length (50 cm effective) to 100 cm total length (90 cm effective) so that it can exit the CE instrument and extend to the MS source. Figure 3 (bottom trace) illustrates the CE–MS electropherograms of desalted, APTS-labelled rMAb glycans using the standard CE–LIF buffer and 100 cm capillary (modified conditions). As expected, an increase in migration time was observed using the longer 100 cm capillary. Most importantly, the overall peak profile remained consistent so identification of the peaks in the CE–LIF profile can be performed by direct comparison with the electropherogram obtained by CE–MS. Mass spectral data obtained for the major peaks confirmed the identity of oligosaccharides with zero, one and two terminal galactose residues (G0, G1 positional isomers and G2, respectively). In addition, the minor peaks were identified as the afucosylated G0 (G0-F) and G0-GlcNAc (G0-1) species.

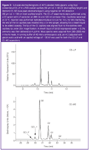
Figure 3
CE in Routine Analysis
CE is commonly avoided in routine analysis because it is reputed to be a troublesome technique with high failure rates. However, this is no longer true because instrument manufacturers have drastically improved instrument design and overall CE knowledge has increased. There are three key contributors to reducing failure rate and producing accurate, precise and robust CE data: Training the operator, system suitablity and instrument maintenance.
Training the operator: General knowledge of the theory of CE, as well as instrument function, is not common amongst analysts. Capillary electrophoresis systems differ significantly in design and operation from HPLC systems and require extensive initial in-depth training. As well as explaining the overall sample preparation steps, such as labelling (CE–SDS–LIF) or sample composition (CIEF), special emphasis should be put into building a cartridge and overall knowledge of capillary coating.
HPLC columns tend to be robust and almost indestructible, whereas coated capillaries can be easily damaged through misuse. Simple errors include the burning of a capillary window by heating wire or applying voltage to a dry capillary. Both of these actions tend to damage the coating, which will result in loss of resolution for protein separation. Uncoated capillaries, such as those used in CE–SDS, tend to be more robust, but special care should be taken with respect to keeping the ends of the capillary clean cut and free of coating because bits of the polyimide coating may clog the capillary during the pressurized gel filling process.
Meaningful system suitability: As well as providing the analyst with the knowledge of science and technique it is also critical to develop a meaningful system suitability test. A system suitability test should not only ensure accurate and reproducible results but should also help the analyst discover underlying issues and help when troubleshooting. For example, besides general ranges on %CPA for the main peak in reduced CE–SDS–LIF, a second criteria can be set with respect to %CPA for incomplete reduced area, which will indicate a poorly performed reduction. To monitor overall separation performance a criteria can be set around migration time precision — because migration time is directly related to the reproducibility of the gel buffer/capillary length/applied voltage.
Instrument maintenance: Most CE instruments are complex and require routine maintenance to reduce failure rates. During the course of a CE separation both the capillary end and the electrode will push through a septum into a vial (sample, separation, wash solution, etc.) multiple times during operation. This mechanical insertion can often lead to bending and breaking of the capillary and/or electrode if the system is misaligned. As the electrode is generally more sturdy than the capillary it is recommended to cut the capillary about 2–3 mm shorter than the electrode and to keep the capillary and the electrode tightly together so that the electrode will guide the capillary through the septa (Note: do keep in mind that both ends of the capillary must be at equal height to reduce gravity-induced flow).
The need for operational care is magnifiied when viscous gels are involved. The vials containing gels are pressurized during the filling process, which often leads to splashing of the gel against vial septa and instrument interface. If left unattended, the gel will dry out and form a thin, conducting film. The presence of this film can result in current spikes, which will affect resolution and create typically non-Gaussian artifact peaks. In severe cases the gel may close off the pressure connection, or create an uneven surface for septa and instrument interface, which will lead to increased pressure failures. It is, therefore, recommended to load the gel with low pressure and to clean the instrument interface block after each session with a wet cotton tip.
Conclusions
Capillary electrophoresis (CE) methods for the analysis of therapeutic proteins are routinely used at Genentech in all stages of product development. These include process and formulation development, physicochemical characterization, comparability and QC lot-release testing. This article highlights the work conducted at Genentech to advance CE applications in the biopharmaceutical industry. First, we optimized the sample preparation scheme for quantitative CE–SDS analysis of non-reduced rMAbs. The inclusion of an alkylation step with 40 mM IAM during heat treatment of non-reduced rMAb samples at 70°C for 5 min in the presence of CE–SDS sample buffers at pH levels of 6.5 significantly suppressed the extent of thermally induced fragmentation and led to increased sample stability. Second, we proved that imaged CIEF is a reproducible technique to assess the charge heterogeneity of therapeutic proteins with the advantages of (1) faster method development, (2) rapid analysis time and (3) the potential of using the same method for a wide variety of rMAbs. We then demonstrated the utility of CE–MS technology to provide accurate mass information on major and minor N-linked glycans of rMAbs. The CE–MS method has been developed with an emphasis on maintaining the same peak profile to that obtained in the CE–LIF assay for more accurate peak identification. Finally, we provided advice and suggestions with respect to CE training and instrument maintenance to ensure high success rates of CE methods in a routine QC laboratory for therapeutic proteins.
Acknowledgements
The authors gratefully acknowledge James McElroy, Yong Liu and Monica Parker respectively for their contributions to the iCIEF, CE–MS and CE–SDS work. In addition, we thank Stacey Ma, Wassim Nashabeh, Dieter Schmalzing and Reed Harris for their support to expand CE technology at Genentech.
Oscar Salas-Solano is a senior scientist in the Protein Analytical Chemistry at Genentech Inc. His work includes physicochemical characterization of proteins, CE technology development, and development and validation of analytical assays for in-process, lot-release and stability testing of recombinant therapeutic proteins.
Lynn Gennaro is a scientist in the Protein Analytical Chemistry at Genentech Inc. Her work includes CE–MS technology development and analytical assays development and validation for the control systems of therapeutic proteins.
Chantal Felten is a consultant at Cardiome Pharma Corp., Canada. Her work focuses on the implementation of CE and HPLC methods for the analysis therapeutic proteins.
References
1. G. Hunt and W. Nashabeh, Anal. Chem., 71, 2390–2397 (1999).
2. M. Schenerman and S. Bowen, Chromatographia, 53, S66-S74 (2001)
3. H. Lee, S. Chang and E.J. Fritsche, J. Chromatogr. A, 947, 143–49 (2002).
4. O. Salas-Solano et al., Anal. Chem.78, 6583–6594 (2006).
5. B. Nunnally, et al., Chromatographia, 66, 955–961 (2007)
6. J. Wu et al., Biotechnol Lab, 17, 24–26 (1999).
7. W.S. Hancock, J. Proteome Res., 1, 297–303 (2002).
8. R. Jefferis, Biotechnol. Prog., 21, 11-16 (2005).
(9) G. Walsh and R. Jefferis, Nat. Biotechnol., 24, 1241–1252 (2006).
10 R. Shields et al., J. Biol. Chem., 277, 26733–26740 (2002)
11. R. Niwa et al., Clin. Cancer Res., 10, 6248–6255 (2004)
12. C. Ferrara et al., J. Biol. Chem., 281, 5032–5036 (2006).
13. G.Walsh, Nat. Biotechnol., 24, 769–776 (2006).
14. R.A.Evangelista, M.S. Liu and F.T Chen, Anal. Chem., 67, 2239–2245 (1995).
15. S. Ma and W. Nashabeh, Anal. Chem., 71, 5185–5192 (1999).
16. J. A. Olivares et al., Anal. Chem., 59, 1230–1232 (1987).
17. J. Henion, A. Mordehai and J. Cai, Anal. Chem.,66, 2103–2109 (1994).
18. J. Axen et al., Electrophoresis,28, 3207–3213 (2007).
19. L.A. Gennaro, O. Salas-Solano and S. Ma, Anal Biochem, 355, 249–258 (2006).















Evaluating Body Odor Sampling Phases Prior to Analysis
April 23rd 2025Researchers leveraged the advantages of thermodesorption, followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC×GC/TOF-MS), to compare and assess a variety of sampling phases for body odor.



