Estimating Uncertainty
LCGC Europe
Estimating uncertainty has become one of the most important metrological concepts in analytical science over the last 15 years to such an extent that some authors consider a result useless or invalid unless it is accompanied with an uncertainty statement. This article describes how to estimate uncertainty in chromatographic analysis and how laboratories can calculate it using data from the method validation process.
Chromatography laboratories must now prove the analytical methods they use provide results that are reliable and fit for the designed purpose.1 Many decisions are taken on the basis of the results provided. Reliability of results is guaranteed if the laboratory assesses the trueness of the analytical method2 and checks it periodically through an established quality assurance/quality control (QA/QC) system.3
Nevertheless, it is also necessary to provide an estimation of the degree of confidence in the results, that is, showing how much a result can deviate from the true value. This means that laboratories must provide accurate results and include an associated uncertainty. Uncertainty is then widely recognized as an essential part of any quantitative analysis.1
Uncertainty is defined as "a parameter, associated with the result of a measurement, which characterizes the dispersion of the values that could reasonably be attributed to the measurand."4 In chemical analysis the measurand refers in many instances to the concentration of an analyte. The term uncertainty means doubt about an analytical result. Therefore, uncertainty gives an idea about the quality of the result because it provides the range of values in which the analyst believes that the "true concentration" of the analyte is situated.
Why is uncertainty so important? As an example, consider two laboratories that analyse the content of aflatoxin B1 in nuts by high performance liquid chromatography (HPLC). Would they report exactly the same result? Probably not. But do they provide comparable results? Uncertainty measurements answer this question. If the two laboratories provide results such as 3.0 ppb and 2.7 ppb of aflatoxin B1, respectively, one cannot indicate whether or not the results are comparable. However, if the results are expressed as 3.0 ± 0.5 ppb and 2.7 ± 0.4 ppb of aflatoxin B1, now one can state both results are comparable (a statistical test would confirm whether or not they are significantly different).
Another example comes from the anti-doping laboratories. The detection in athletes' urine of the metabolite 19-norandrosterone, in amounts above 2 ng/mL, constitutes an adverse analytical finding. However, does a reported level of 2.3 ng/mL of 19-norandrosterone means a positive result? The answer is that one does not know. The decision limit (L) follows by considering the uncertainty (U) in the measurement value at the threshold level (T) of 2 ng/mL because this level is the highest level that complies with the null hypothesis, that is, the athlete in question is "clean''. Thus only a result above the decision limit — L = T + U — can be declared as positive.5
Uncertainty and traceability are closely related concepts. If the traceability of the analytical method is not assessed, one cannot guarantee the correction of all the possible systematic errors. Consequently, it is impossible to ensure that the "true value" is included within the interval "value 1 ± value 2" (where value 1 is the best estimation of the measurand obtained when analysing the sample and value 2 is the uncertainty of value 1). Therefore, as Figure 1 shows, every analyst should verify the trueness of the method before calculating uncertainty.



Figure 1
Estimating Uncertainty
Uncertainty can then be estimated using the information generated in the method validation process and, specifically, in the assessment of trueness. Moreover, precision and robustness studies also give useful information for calculating uncertainty.6,7 The uncertainty calculated using this information can consequently be related to the results of routine test samples as long as the reference sample is representative for these test samples and the QA conditions are implemented in the laboratory.
There are four main steps in the uncertainty estimation process, as described in the EURACHEM/CITAC Guide and shown in Figure 2.7

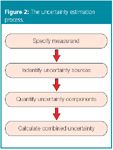
Figure 2
First, a clear statement of what is being measured, including a quantitative relationship (e.g., in the form of an equation) between the measurand and the input quantities upon which it depends, has to be written down. This specification information should be given in the relevant standard operating procedure (SOP) or other method description.
The second step is to list the possible sources of uncertainty for the analytical procedure. For example, different operators, environmental conditions (such as temperature or pressure), calibration or integration techniques cause variability in the chromatographic results. In gas chromatography (GC) there are other or additional factors that may affect the results. For example, the column temperature, the carrier gas flow-rate, the injection temperature, the split ratio and the sample size. In liquid chromatography (LC), a major source of error in the peak height and peak area measurements is associated to the detector noise because of flow-rate fluctuations. An extensive list of uncertainty sources in chromatographic analysis can be found in reference 8. A general procedure for forming a structured list of sources is to use so-called cause-and-effect diagrams (Figure 3).7
The third step is the quantification of these sources of uncertainty. Different possibilities exist. In a first approach, called the error-budget or bottom-up approach, the uncertainty caused by each of the identified sources is quantified and then combined. However, the individual estimation of all contributions to uncertainty is far from evident, especially when many sources can be identified.
As an alternative, it is often possible to estimate a contribution to the uncertainty associated with those from a number of separate sources. For example, the overall method precision usually accounts for a significant proportion from several sources of uncertainty. For this approach, the laboratories can use historical data that is already available such as (1) data from validation studies, such as assessment of trueness and precision studies; (2) data from internal QC procedures; or (3) data from method collaborative studies or proficiency testing schemes. The latter approach also is called the top-down approach. A comparison of different approaches to estimate the uncertainty of a liquid chromatographic assay is made in reference 9. The main disadvantage of the alternative approaches is that uncertainty is estimated globally and not individually for different sources of variability. Therefore, the analyst has little information on individual sources to try to improve the chromatographic method and reduce the uncertainty.
It is important to consider if the available data sufficiently accounts for all sources of uncertainty and occasionally plan additional experiments and studies carefully. Robustness studies, for example, can be used if there are sources of error that have not been representatively varied during the precision studies.9
The information obtained in the previous step will consist of a number of quantified contributions to overall uncertainty, either associated with individual sources or with the combined variability of several sources. The contributions are expressed as standard deviations and combined according to the appropriate rules (of error propagation), to give a combined standard uncertainty, u. An appropriate coverage factor, k, is finally applied to give the so-called expanded uncertainty, U, which provides an interval within which the value of the measurand is believed to lie with a certain level of confidence (typically 95%).
Equation 1 shows a typical expression for the expanded uncertainty, here calculated as the sum of four terms. However, because uncertainty can be considered under different circumstances (within-laboratory uncertainty, reproducibility uncertainty,...) and the contribution of different sources of variability can be estimated differently, many alternative equations can be found in the literature.9,10


The first two terms, that is, the uncertainty of the analytical procedure, u rel,proc , and the uncertainty of the assessment of trueness, u rel,trueness , can be calculated from the information generated in the assessment of trueness. The uncertainty of the pre-treatment steps, u rel,pret , is necessary when the sources of uncertainty arise from subsampling and from preprocessing steps applied to the routine samples and which have not been considered in the assessment of trueness or precision studies to estimate u rel,proc . Finally, the uncertainty of other terms, u rel,other , takes into account all the sources of uncertainty that have not been considered in the preceding terms. For example, the differences in the recovery depending on the sample matrices and/or analyte concentrations.


Figure 3
The subscript rel means the uncertainty components are expressed as relative standard uncertainties. In this way, the different components can be added, regardless of the concentration level at which they have been estimated. The final combined relative standard uncertainty (square root in the equation) is multiplied by the concentration, c, obtained from the chromatographic analysis of the sample, to get an absolute combined uncertainty. In this way, uncertainty is assumed to be proportional to concentration. Finally, a coverage factor of k = 2 is usually applied to have a level of confidence of about 95%.
Uncertainty of the analytical procedure, u rel,proc : This uncertainty corresponds to the intermediate precision of the method (i.e., the precision obtained within a laboratory by varying all the factors that can affect the chromatographic measurement: day, operator, instrument, calibration, etc.). Intermediate precision is usually obtained from precision studies. However, QC data and trueness studies can also be used to estimate this precision.
If the chromatographic method is to be applied in a wide concentration range, precision should be determined at three levels of concentration (i.e., at low, medium and high). Then, the standard deviation (SD) and the relative standard deviation (RSD) have to be calculated. The intermediate precision is proportional to concentration if there are no significant differences between the RSD for each level of concentration. In this instance, the RSD can be pooled to give a single estimation of the uncertainty of the procedure:

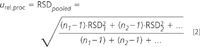
where RSD1 is the relative standard deviation calculated for the sample at concentration 1, n1 is the number of replicates for that sample, and so on.
For wide concentration ranges, it is not unusual to find that precision is not directly proportional to concentration over the whole concentration range. In this instance, the uncertainty of the procedure should be calculated for the individual concentration ranges or modelled as a function of the concentration.
Uncertainty of trueness, u rel,trueness : This component derives from the fact we are uncertain about the bias of the method (checked when assessing trueness). The best way to assess trueness (the one providing a higher level of traceability) is to use a certified reference material, CRM.2 However, CRMs are seldom available for many types of matrices and levels of concentration required in chromatographic analyses. Therefore, trueness is usually assessed with spiked samples, through recovery studies. It is important to note that before calculating uncertainty, trueness always has to be assessed. Otherwise, it cannot be guaranteed that the "true" concentration value lies within the interval Result 1 ± Result 2 with a given probability (usually 95%).
In recovery studies, the reference sample has to be analysed under intermediate precision conditions at least seven times.2 If the uncertainty of the reference value is negligible and the recovery does not differ significantly from one, the uncertainty of recovery is calculated as:

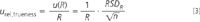
where R is the recovery and RSDR is the relative standard deviation of the n results obtained when analysing the reference sample. This RSDR is also an estimate of the intermediate precision because the sample is analysed under intermediate precision conditions. Therefore, it could also be used to estimate the component of uncertainty as a result of the analytical procedure, u rel,proc (see Equation 1).
Trueness also has to be assessed to at least three levels of concentration (low, high and medium) if the chromatographic method is to be applied in a wide concentration range. An average recovery (together with its uncertainty) can then be calculated for the whole concentration range if there are no significant differences between the levels of concentration.
Uncertainty of pretreatment steps, u rel,pret : The uncertainty arising from heterogeneity and/or pre-treatment steps can be calculated from a sample with the same characteristics as the routine working samples. The sub-sampling and/or the preprocessing must be performed by representatively changing all the factors that may affect them. Then, the chromatographic analysis of each portion sub-sampled and/or pre-treated should be performed under repeatability conditions. In this way, the uncertainty of the pre-treatment steps is not overestimated because the variability of the repeatability of the chromatographic analysis can usually be neglected.
Uncertainty of other terms, u rel,other : This uncertainty takes into account sources of variability that have not been representatively varied during the precision studies. These sources of uncertainty can be estimated using the information from robustness studies or from historical information already available by the laboratory. An example is the component of uncertainty because of the calibration of a balance. If the estimation of the intermediate precision of the analytical procedure has been performed within one single calibration, then this component is not included in the precision estimate and should be added using information from the calibration certificate of the balance.
This uncertainty can also be a result of the variability of recovery with the sample matrix and/or analyte concentration. In this instance, the analyte has to be spiked to different representative sample matrices and/or at several levels of concentration. The recovery for each spiked sample should be calculated by analysing all the samples under repeatability conditions. The uncertainty of the matrix/sample variability corresponds to the standard deviation of the recoveries.
Conclusions
This article discusses the importance of estimating measurement uncertainty in chromatographic analysis and describes a possible way to estimate this uncertainty. Once the measurand is specified, the different sources of uncertainty are identified. Then, the uncertainty can be estimated in a global way by grouping the previously mentioned sources of uncertainty in a few components. By grouping, the calculation of uncertainty becomes easier and enables the laboratory to use (already available) historical information, obtained during the validation process.
Ricard Boqué is an associate professor and Alicia Maroto a post-doctoral researcher at the Universitat Rovira i Virgili, Tarragona, Spain, working on Chemometrics and Qualimetrics. Yvan Vander Heyden is a professor at the Vrije Universiteit Brussel, Brussels, Belgium, department of Analytical Chemistry and Pharmaceutical Technology, and heads a research group on Chemometrics and Separation Science.
References
1. ISO/IEC 17025:2005. General requirements for the competence of testing and calibration laboratories. International Organization for Standardization, Géneve, Switzerland.
2. R. Boqué, A. Maroto and Y. Vander Heyden. LCGC Europe, 21, 264–267 (2008).
3. M. Thompson and R. Wood, Pure Appl. Chem., 67, 49–56 (1995).
4. ISO Guide 99:1993. International vocabulary of basic and general terms in metrology (VIM). International Organization for Standardization, Géneve, Switzerland.
5. N.M. Faber and R. Boqué, Accred. Qual. Assur., 11, 536–538 (2006).
6. A. Maroto et al., Anal. Chim. Acta., 440, 171–184 (2001) .
7. EURACHEM/CITAC Guide. Quantifying Uncertainty in Analytical Measurement, 2nd ed., 2000.
8. V. J. Barwick, J. Chromatogr. A, 849, 13–33 (1999).
9. E. Hund, D.L. Massart and J. Smeyers-Verbeke, Anal. Chim. Acta., 480, 39–52 (2003)
10. E. Hund, D.L. Massart and J. Smeyers-Verbeke, TRAC-Trend Anal. Chem., 20, 394–406 (2001) .



















