HPLC and UHPLC Columns: Then, Now, Next
LCGC North America
In advance of Pittcon 2018, leading scientists-Ronald Majors, Richard Henry, John W. Dolan, Zachary S. Breitbach, and Daniel W. Armstrong-who will be speaking at the LCGC awards symposium give us a preview of their talks.
In advance of Pittcon 2018, leading scientist Ronald Majors, who will be speaking at the LCGC awards symposium, gives us a preview of his talk.
The column has always played a pivotal role in the capability of liquid chromatography (LC) to solve "real world" problems. From the early days of large particle superficially porous packings (SPPs) to the eventual decades of domination of 5-µm reversed-phase columns to today's modern monolithic, sub-2-µm porous and the (revisited) small-particle SPPs, the state of column technology has often led, and sometimes fallen behind, the capabilities of the instrumentation that it supports. My presentation at the conference will highlight the developments in high performance liquid chromatography (HPLC) (and the newer term ultrahigh-pressure liquid chromatography [UHPLC]) columns that have brought liquid-phase separations to an elevated level of contribution to the analytical laboratory. We will also look into our crystal ball and delve into possible future directions in column technology.
Early Developments
The birth year of HPLC is generally recognized as 1966 (1). Then, microparticulate ion-exchange resins (10 µm diameter), SPP (45–60 µm), and flow-through ultraviolet (UV) detector development capped a year of discovery. To follow these and future developments, Figure 1 shows pivotal events in the development of column particle technology from the early 1960s onward. For most practitioners, the development of SPPs was a breakthrough since separations previously performed on large porous silica particles could now be accomplished in a fraction of the time. The SPPs were rather large, consisting of a glass bead or solid-core silica as the base material and a thin porous layer (thickness 1–2 µm) as the separation media. Relative to the larger (greater than 100 µm), totally porous particle (TPP), column efficiencies were quite good, but the low surface area limited the sample capacity of the packed column. The improvement in efficiency was because of the short diffusion pathlength in this thin porous layer. Nevertheless, in the early 1970s, SPPs were the rage. They could be dry-packed by the user much like a packed gas chromatography (GC) column of the day.
Another critical development in the early 1970s was the advent of bonded stationary phases. The older coated phases were very unstable and frequently stripped off the surface of the SPP. The mobile phase had to be saturated with dissolved stationary phase, which was very inconvenient and almost impossible to run any type of gradient elution. The bonded phases (monomeric and polymeric) allowed the use of more-polar solvents and gave way to the development of reversed-phase chromatography, which eventually came to dominate the mode usage and carries on today.
It had long been known, based on the decades earlier work of Martin and Synge (2), that "the smallest HETP (height equivalent to a theoretical plate) should be obtainable by using very small particles and a high-pressure differential across the length of the column." In the late 1960s, there were no sub-10-µm silica particles available-that is, until E. Merck (Darmstadt, Germany) was able to fractionate their irregularly shaped, thin-layer-grade silica into smaller particle sizes. Using a balanced density slurry procedure (slurry packing techniques are still used today), stable and reproducible silica gel columns were packed with 5–10 µm particles and demonstrated much higher efficiencies than the SPP columns (3). Figure 2 shows the first chromatogram obtained with a synthetic mixture in 1971 (3). This chromatogram looked pretty good, even by today's standards. After the 1971 commercialization of sub-10-µm TPP columns, the developments in column technology took off with spherical porous silica particles, a wide variety of siloxane bonded phases for several modes of HPLC, wide-pore packings for biomolecules being matched by the development of high-pressure HPLC instrumentation, and a variety of detectors and data systems that could quickly measure peak areas, retention times, and provide easy-to-read chromatographic outputs.
Now
Referring again to Figure 1, as the years rolled on, the particle size of HPLC packings continued to decrease each time resulting in a doubling of column efficiency and speed. Speed was enhanced by the proportional decrease in column length to maintain the same efficiency.



Figure 1: History of commercial HPLC particle development.
For most of the 1980s and into the 1990s, workers seemed to be content with the use of 5-µm TPP columns (Figure 3a) and instruments capable of 400-bar operation. Smaller particle size nonporous-resins and silicas came about, but because of very low sample capacity and frequent pressure problems they quickly fell by the wayside. Gradually, 3–3.5 µm particles found use in some analytical laboratories but in the new millennium, 1.8-µm packed columns (subset of Figure 3a) were introduced, accompanied by a new range of instrumentation with a new generation of pumps to work with these higher-pressure columns. The pressure race was on. However, narrower peaks and the anticipation that TPP sizes would continue to decrease led manufacturers to also focus on smaller extracolumn volumes to handle the narrower peaks from these columns. The resultant technique became UHPLC, which implied that chromatographers had to upgrade their existing instruments to perform better with these columns.


Figure 2: First successfully packed HPLC column using microparticulate (5–10 µm) silica, circa 1971 (3). Column: 15 cm × 2.1 mm; mobile phase: n-hexane; flow rate: 400 mL/h; detection: UV absorbance; temperature: ambient; sample: 4 µL of mixture of ~1 mg/mL of each component. Peaks: 1 = impurity, 2 = phenetole, 3 = nitrobenzene, 4 = methyl benzoate, 5 = carbazole, 6 = acetophenone, 7 = 2,4-dinitrobenzene.
Note from Figure 1, that along the way, the concept of SPPs resurfaced, mainly driven by the work of the late J.J. Kirkland (4), who was a proponent of SPP in the early days. However, this time the size of the solid-core material was more than an order of magnitude smaller than the SPPs of the 1960s. The shell was thinner. There were also some new particle names: fused-core, core–shell, solid-core, and Poroshell, to name a few. Kirkland's SPP introduction was for the high-speed separation of large biomolecules such as proteins. Because of their size, proteins diffuse very slowly in solution, with about 10 times lower diffusion coefficients than those typical of small molecules. Therefore, as shown in Figure 3b, the short diffusion pathlength of the wide-pore packing brought about many advantages compared to totally porous particles of the same size, allowing large molecules to diffuse into and out of this thin layer very quickly, even at relatively high flow rates, thereby improving mass transfer kinetics. The larger 300-Å pore size was large enough to accommodate proteins up to 500 kDa.

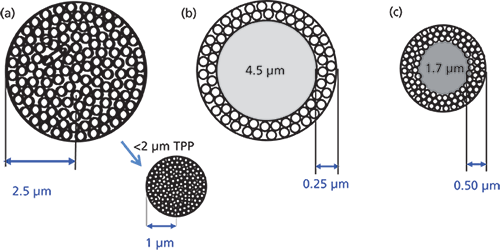
Figure 3: Comparison of various particle types: (a) 5-µm totally porous particle (TPP), (b) 5-µm superficially porous particle (SPP), and (c) 2.7-µm SPP.
However, the biggest breakthrough for small-molecule separations came in the late 2000s when Kirkland once again brought the SPP technology to the forefront (5). By their introduction, the sub-3-µm SPP particles generated a significant interest in the chromatographic community. A typical example of the profile of a 2.7-µm SPP particle is shown in Figure 3c. This configuration has a 1.7-µm solid core with a 0.5-µm silica shell and 120-Å pores. These particles gave rise to column efficiencies rivaling the sub-2-µm TPP but, because of their 2.7-µm particle size, the pressure drop for the same size column was approximately 40–50% lower. Thus, in addition to the higher-pressure UHPLC instruments, most SPP columns of this particle size can be used with some of the conventional 400-bar HPLC systems. Longer columns can be used to generate more theoretical plates with UHPLC systems for more difficult separations. The number of sub-3-µm commercial SPP products took off like a rocket, and they appear to be replacing the sub-2-µm TPP, albeit with much lower operating pressures while operating at the same efficiency with only a slightly reduced sample capacity. Even smaller SPP columns (down to 1.3 µm) are now available. Needless to say, SPPs are here to stay and hardly a whisper is heard about sub-2-µm TPPs. A comparison of the two types of columns is shown in Figure 4.

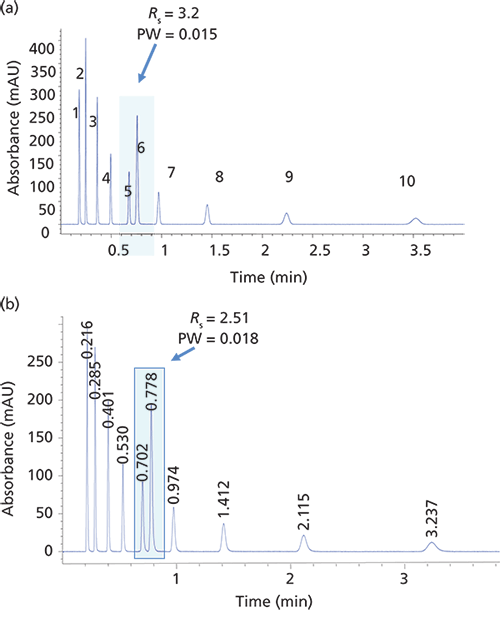
Figure 4: Isocratic separation of alkylphenones on (a) a totally porous particle column and (b) a superficially porous particle column. Column: (a) 50 mm × 4.6 mm, 1.8-µm Zorbax Eclipse Plus C18, (b) 50 mm × 4.6 mm, 2.7-µm Poroshell 120; pressure: (a) 358 bar, (b) 220 bar; mobile phase: 60:40 acetonitrile–water; flow rate: 2.5 mL/min. Peaks: 1 = uracil, 2 = acetanilide, 3 = acetophenone, 4 = propiophenone, 5 = butyrophenone, 6 = benzophenone, 7 = valerophenone, 8 = hexanophenone, 9 = heptanophenone, 10 = octanophenone.
One other current column type will be addressed: Where do monolithic columns fit into the chromatography laboratory? Although monoliths have been around for more than two decades, they have not come close to replacing microparticulate columns. Research continues to provide improved performance for both silica- and polymer-based monoliths. So, they shouldn't be counted out. Figure 5 shows that they may outperform SPP in the long run.

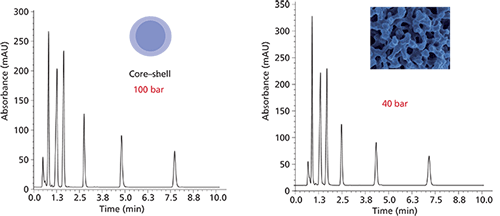
Figure 5: Separations obtained using a second-generation silica monolith column (left) and a superficially porous particle column (right). Columns: (left) 50 mm x 4.6 mm monolith, (right) 50 mm x 4.6 mm, 2.6-µm core–shell; mobile-phase A: 20 mM phosphate buffer, pH 4.5; mobile-phase B: acetonitrile; gradient: 20–40% B in 12 min; flow rate: 1 mL/min; injection volume: 2 µL; detection: UV absorbance at 230 nm. Peaks: 1 = ascorbic acid, 2 = 4-hydroxybenzoic acid, 3 = benzoic acid, 4 = sorbic acid, 5 = methyl-4-hydrobenzoate, 6 = ethyl-4-hydroxybenzoate, 7 = propyl-4-hydroxybenzoate. (Courtesy of Merck Millipore.)
Next
The concluding part of this presentation will briefly look at possible future directions for column technology, including improved monoliths; microbore, capillary, and microfluidic platforms; engineered (ordered) columns via micelle templating with pseudomorphic transformation; pillar arrays; and the potential application of three-dimensional (3D) printing techniques. With regards to selectivity and peak capacity improvements, two-dimensional (2D) (and even 3D column configurations) chromatography has received lots of attention and with modern instrumentation now available, multidimensional techniques may become another daily tool in the liquid chromatographer's arsenal.
For a more detailed expansion of the themes introduced here, please consult references 1, 6, and 7.
References
(1) R.E. Majors, LCGC North Am. 33(11), 818–840 (2015).
(2) A.J.P. Martin and R.L.M Synge, Biochem. J. 35, 1358–1368 (1941).
(3) R.E. Majors, Anal. Chem. 44, 1722 (1972).
(4) J.J. Kirkland, Anal. Chem. 64, 1239–1245 (1992).
(5) D.S. Bell, LCGC North Am. 33(6), 386–395 (2015).
(6) R.E. Majors, LCGC North Am. "The Best of Column Watch and Sample Prep Perspectives: A Farewell to Ron Majors," 33(s11) (2015).
(7) R.E. Majors, LCGC North Am. 33(12), 886–893 (2015)

Ronald E. Majors

Ronald E. Majors is an analytical consultant in West Chester, Pennsylvania and the recipient of LCGC's 2018 Lifetime Achievement in Chromatography Award.












; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")


Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.

, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

