Historical Developments in HPLC and UHPLC Column Technology: The Past 25 Years
LCGC North America


During the course of my scientific career beginning in the 1960s, I have grown up with the birth of modern LC column technology, the refinements of the instrumentation, and the development of widespread application of this most powerful separation and analysis technique. In this installment, I would like to share with you some of my observations and experiences with the beginning, the growth period, and the maturation of HPLC columns, where I have focused nearly 33 years of writing for this magazine. I will explore some of the early column breakthroughs beginning with the development of large superficially porous particles (SPP), the porous irregular and spherical microparticulate particles, inorganic and organic polymeric monoliths and the rebirth of the current generation of SPP. In next month’s installment I will look into my crystal ball and see what the future of HPLC and UHPLC holds.
During the course of my scientific career beginning in the 1960s, I grew up with the birth of modern liquid chromatography (LC) column technology, refinements of the instrumentation, and the development of widespread application of this most powerful separation and analysis technique. Here, I would like to share some of my observations and experiences from the beginning, the growth period, and the maturation of high performance liquid chromatography (HPLC) columns, where I have focused nearly 33 years of writing for this magazine. I explore some of the early column breakthroughs beginning with the development of large superficially porous particles (SPPs), the porous irregular and spherical microparticulate particles, inorganic and organic polymeric monoliths, and the rebirth of the current generation of SPPs.
As high performance liquid chromatography (HPLC) approaches its 50th anniversary, it is appropriate to look back at its beginnings and understand the revolutionary breakthroughs that occurred as well as the ongoing evolution of this powerful separations technique that has had a profound effect on analysis in virtually every field of chemistry.
In my years of writing for LCGC, like the magazine, I have tended to focus on the practical side of the technology avoiding esoteric research and highly theoretical treatments of the science. Instead, I focused on the commercial side, not only because I have been on that side of science throughout most of my career, but because presenting techniques that can be used and products that can be obtained today to solve real problems and not a decade down the road (or perhaps never) is more interesting and satisfying to me.
In July 1994, I wrote an LCGC article entitled “Twenty-Five Years of HPLC Column Development-A Commercial Perspective” (1), which is reproduced in the special supplement included with this issue (2). In that article, I focused on those column and instrument developments that were made, nearly from the beginning of LC, providing the readers with products that they could buy to immediately begin performing HPLC with the state-of-the-art tools of the time. Rather than repeating the details of that original article (1,2), here I focus on the LC technology that has happened in the past 25 years. Instead of covering all aspects of HPLC development (for example, instruments, data systems, automation, and so on), I focus on the separation column and its stationary phases that, in my opinion, have always been the most important part of the HPLC system and have been a major subject in my 33 years of coverage first in LC and later LCGC magazine. The column is where the separation occurs and if the wrong column or separation conditions are chosen, no matter how sophisticated or expensive the instrument that is used, the chromatographic experiment will fail. The column has defined the instrumentation and, at times, vice versa, but instrumentation development has always had a greater lag time than column and chemistry development.
Our ultimate goal in HPLC is to obtain the best possible resolution for the target compounds in the shortest possible time. In column technology, there are two important areas that affect resolution that have changed dramatically from the very beginning: column particle size or format and stationary-phase chemistry. The column particle size or domain size affects column efficiency while the stationary phase affects the other two elements of the resolution equation: selectivity and retention. We look at both of these areas in some detail here.
This month, I look at the continuing efforts that defined HPLC and led to ultrahigh-pressure liquid chromatography (UHPLC) and then take a look at the major practical contributions that have moved the technology into its current practice, Next month, I will look into the crystal ball and glance at possible future directions.
When Did HPLC Begin?
The following question arises: What was the pivotal event (or perhaps series of events) that caused LC to break away from its older practice and become a technique that chemists and biologists seriously considered for their analytical results? Before I get into the details, I would like to look back at the beginnings of what eventually became HPLC. In my July 1994 article (1,2), I loosely defined the research of the late Csaba Horváth, who as a graduate student in the laboratory of István Halász developed the concept of a pellicular particle (now called a superficially porous particle [SPP]) around 1968, as the historical year of the foundations of HPLC. However, looking into it deeper and reading a well-written chapter on Milestones in the Development of LC by Lloyd Snyder (3), I found two earlier publications that to me were the pivotal works that followed the monumental prediction of Martin and Synge in 1941 (4) that clearly stated that “. . . the smallest HETP (height equivalent to a theoretical plate) should be obtainable by using very small particles and a high pressure difference across the length of the column.” That statement resembles the current practice of LC.
The first is a little known 1966 paper by Piel (5) written as a short contribution to Analytical Chemistry where the author slurry-packed finely ground silica, calcium carbonate, or alumina particles into 1- or 2-mm i.d. glass columns. The mobile phase was driven by centrifugation or with a high liquid pressure differential using a pump capable of 3500 psi operation. The particles used varied from less than 1-µm to 0.012-µm. The centrifugal driving force was 1000-1500 times gravity and was applied for 5 min while the pump’s full pressure was applied. The beds varied from 1.6-4.0 cm in length. Samples included various dyes as well as a spinach extract. Separations took only a few minutes for both operations. Piel claimed “excellent resolution and high capacity of microparticulate beds” and showed the results in his accompanying figures.
The second paper that was a precursor to HPLC was that of Hamilton (6), also published in 1966 and applied to amino acid analysis using a pump to drive gradient solvent mixtures through the analyzer. In his work, Hamilton used 10-µm ion-exchange resins. Not only did the use of the smaller narrow particle-size distribution resin give more rapid separations compared to earlier work in this area, but Hamilton’s studies also gave insight into the effect of particle size on efficiency and phase chemistry on selectivity, things we still talk about today.
Thus, if one is considering the birth of HPLC, 1966 sounds like a very pivotal year to note. And this tells us that in 2016 we should celebrate the 50th year of HPLC, its Golden Jubilee.
Column Efficiency in HPLC Totally Porous Particles
Let us first consider the impact of column particle size on efficiency. Table I provides a simple pictorial and chronological overview of the evolution of particle size and technology during the history of HPLC. The above-cited paper of Martin and Synge in 1941 (4) predicted the use of small particles to improve efficiency. However, particles for adsorption and modes of chromatography in popular use today were not available in the small particle sizes that Martin and Synge may have envisioned for liquid-partition chromatography. For at least 25 years, chromatographers used large (greater than 100-µm) porous particles of silica gel and alumina to perform their separations, usually in open glass columns with gravity solvent feed. These large particles gave poor chromatographic performance and slow separations. Attempts to speed up the separation by applying pressure to the system resulted in even broader peaks. Flow-through detectors were not readily available so users would collect fractions and measure them spectroscopically or by other analytical techniques and construct a chromatogram, albeit a time-consuming and error-prone process.
As indicated earlier (1,2), the first real breakthrough in column technology came in the late 1960s when Professor Csaba Horváth of Yale University came up with a support with an impenetrable, spherical solid-core (a glass bead, 37-50 µm in diameter) coated with an outer layer of porous solid (thickness 1-2 µm carbon black or polymeric coating with ion exchange functionality) that provided an order of magnitude improvement in efficiency compared to the older large porous particles. The original work for liquid-phase separations was to be applied to macromolecules such as nucleic acids and their derivatives. Horváth and coworkers (8) demonstrated the pellicular particle (also called at the time porous layered bead [PLB] for silica particles or SPP in general) for the ion-exchange separation of nucleosides. The gain in efficiency was attributed to the thin porous layer that restricted the diffusion of large molecules into deep pores, typical of larger porous particles. These dense SPPs, usually dry packed by the user into 50- or 100-mm columns of 2.0-mm internal diameters, were an immediate success; commercialization brought these products into the hands of chemists who were now able to solve “real world” problems in a matter of tens of minutes instead of hours. Since the particles were smaller than those used earlier, a high-pressure pump was required to push mobile-phase solvent through a packed bed; hence, the name high pressure liquid chromatography became synonymous with the use of these new columns. Later high performance LC became more acceptable.
I would be remiss if I did not further mention the work of Horváth and Sandy Lipsky, also a Yale professor, (8) who built the first liquid chromatograph capable of high-pressure operation that also had a low dead-volume ultraviolet (UV) absorption detector. They were not engineers in the sense of instrument design and the chromatograph had a number of deficiencies (such as electrical lines and solvent lines running next to each other and excessive dead volumes) but, nevertheless, it was a unit capable of providing decent gradient chromatograms using their pellicular ion exchange packing in small-diameter columns (1-mm i.d.). The unit was commercialized specifically as a nucleic acid analyzer and for the first time (late 1960s), a chromatographer could purchase an LC system ready to use instead of having to assemble it by oneself or trying to adapt a commercial amino acid analyzer or size-exclusion chromatograph to general LC. This early HPLC instrument was followed by well-engineered, more general purpose systems from Waters, DuPont, Varian, and others and the explosion of HPLC began. For the complete story of the early SPP, the reader is referred to the first 25 years article from 1994 (1,2).
Although the SPP columns were a major breakthrough, considered by most to be a revolutionary development in liquid chromatography separations, and the dense particles could be dry-packed by the user, the thin layer of porous material provided a low surface area (approximately 2-4 m2/g) or ion-exchange capacity compared to the larger porous particles (300-400 m2/g). Thus, when using columns with these particles for preparative work or with low sensitivity detectors such as the refractive index detector, a larger concentration of sample had to be injected and the sample capacity of these materials proved to be quite low to handle these larger masses of sample. More surface area was needed and thus the search was on for another solution.
So, further work on obtaining smaller totally porous particles (TPP) of silica gel to fully realize the predictions of Martin and Synge continued. In the early 1970s, larger quantities of small size porous particles became available with the advent of air centrifugal classifiers. Attempts to dry pack these smaller porous particles below 20 µm in diameter were futile. As depicted in Table I, the next breakthrough came in 1971 when balanced-density slurry techniques were used (9) that enabled the successful packing of 5-10 µm porous irregularly shaped silica gel particles into narrow-bore columns (2-mm i.d.). These columns were commercialized as MicroPak columns in 1972. Now chromatographers had columns that provided even higher efficiency than the early SPPs and achieved high sample capacity. The irregularly shaped porous silica particles first used gradually gave way and were replaced by spherical silicas that packed more uniformly. Again, readers are directed to the original reference to hear more about the strides made in TPPs (1,2).



As depicted in Table I and demonstrated pictorially in Figure 1a and chromatographically in Figure 2, over the next three decades, porous spherical microparticles continued to decrease in size resulting in more efficient columns. HPLC became an established separation technique, especially for the separation of nonvolatile, ionic, ionizable, and polymeric compounds, and just about every laboratory doing chemical analysis had one or more liquid chromatograph at their disposal. For columns of equal dimensions, each time a decrease in particle size was achieved, column pressure increased with the inverse square of the decrease in particle diameter and high-pressure pumps with 400-bar capability were required. Fortunately, the increase in efficiency allowed shorter columns to be used and 6000-psi (400-bar) pumps and systems were the standard for HPLC laboratories over this time period.


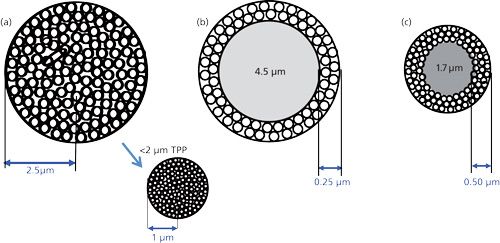


By 1995, spherical silicas dominated the HPLC marketplace with irregular particles mainly relegated to preparative LC because of their lower cost. Although 5-µm particles were the most widely used for analytical LC, 3.0-3.5 µm particles were gaining attention. These particles were packed into shorter columns (for example, 15 cm versus 25 cm for 5-µm particles), but gave equivalent separations to the longer columns packed with 5-µm particles. The 10-µm particles of yesteryear were still the most popular for preparative LC. Mass spectrometry (MS) was becoming a more standard method of detection for HPLC but it needed lower flow rates than the popular 4.6-mm i.d. columns offered. These users tried some of the shorter columns (50 mm in length) with smaller diameters (down to 2.1 mm) that gave lower flow rates, but they were already experiencing some difficulties with extra column effects of their instruments. Those playing with packed fused-silica columns and microbore columns (1-mm i.d. or so) had similar problems.
However, in the mid-1990s, even smaller particle size packings came onto the market. The nonporous particles of 1.5-µm diameter, designed for biomolecules for fast separations, showed excellent efficiencies but gave rise to column head pressures that exceeded the capability of existing pumping systems. The nonporous resins and nonporous silicas were rather short-lived since they often plugged, had extremely low capacity, and instruments weren’t really suitable to provide widespread application. Some of these columns were tried for small molecules but they also went by the wayside.
For most of the 1990s and early 2000s, workers were content to live with available columns and focused on the myriad of chemistries that were introduced during this time period (see later section on stationary phase developments). The next big area of focus occurred in 2003, when Agilent Technologies introduced the 1.8-µm totally porous columns Zorbax Rapid Resolution at Pittcon (10) thereby breaking the 2-µm barrier. As depicted in Table I, this was an evolutionary direction in column technology, as smaller particle sizes lived up to the original Martin and Synge (4) prediction. The introduction of the Acquity UPLC (Ultra Performance LC) system by Waters a year later gave higher pressure capability to the marketplace than the conventional 400 bar (6000 psi) and the pressure race was on. More commonly referred to as ultrahigh-pressure liquid chromatography (UHPLC), a term coined by Jim Jorgensen of the University of North Carolina, it eventually led to pumping systems capable of pressures up to 19,000 psi.
However, throughout the development of LC, researchers experimented with even smaller particles and higher pressures beginning with the work of Bidlingmeyer and Rogers (11,12) and later with work of Jorgensen and colleagues (13). The latter provided some convincing separations using nonporous and porous particles as small as 1 µm and pressures as high as 80,000 psi. However, in the real world operating at such high pressures has not proven suitable for routine use since physiochemical solute and mobile-phase parameters are affected by pressure, safety may be compromised, and instrument manufacturers have not endorsed operating at extremely high pressures. More recently, the work of Wirth and coworkers (14) using submicrometer particles for the separation of proteins employing slip-flow characteristics has shown some phenomenal efficiencies and the jury is out on whether these now commercial columns will establish themselves as mainstream.
Beginning in the mid-2000s, each year more and more sub-2-µm totally porous particle (TPP) columns became commercially available; my last count tallied more than 30 companies with such columns in their portfolios. For my last published listing, see reference 15.
However, when more difficult separations are encountered, sub-2-µm particles are packed into longer columns of 100-150 mm; then, at normal flow rates (~2 mL/min) and typical mobile phases, pressures exceed the 400-bar capability of most commercial systems; thus UHPLC systems were recommended. Initial UHPLC systems started at 6000 psi and slowly those capable of operation up to 19,000 psi came onto the market, allowing the use of these smaller particle columns for higher speed and high efficiency separations. Most users have not operated their instruments anywhere near these maximum operating pressures, but it is nice to know that one has the capability if needed. A fallout of the pressure race has been that the current UHPLC systems also have reduced band dispersion performance which allows these sub-2-µm TPP columns to come closer to their full level of efficiency. Nevertheless, in recent years, fewer sub-2-µm TPP products have been introduced and none are below 1.5 µm. This reduction in further interest is undoubtedly because of the widespread acceptance of a new generation of SPPs.
Superficially Porous Particles
An important feature of Table I to point out is that the SPP concept has been resurrected in recent years. However, this time the size of the solid-core material is more than an order of magnitude smaller than the pellicular packing of the past. The shell is also thinner. There are also some new particle names: fused-core, core-shell, and poroshell, to name a few. The first of the new breed of SPP was introduced by Kirkland (16) as a packing material for the high-speed separation of large biomolecules such as proteins. Due to their size, proteins diffuse very slowly in solution, about 10 times lower diffusion coefficients than those typical of small molecules. Therefore, a short, diffusion path length brought about many advantages compared to totally porous particles of the same size. Figure 1b shows that the particle size of the Poroshell 300 (Agilent Technologies) averaged 5 µm while the phase thickness was only 0.25 µm. Since they cannot diffuse into the solid core, proteins and other large molecules diffuse into and out of this thin layer very quickly, even at relatively high flow rates, thereby improving mass transfer kinetics. The larger 300-Å pore size is large enough to accommodate proteins up to 500 kDa.
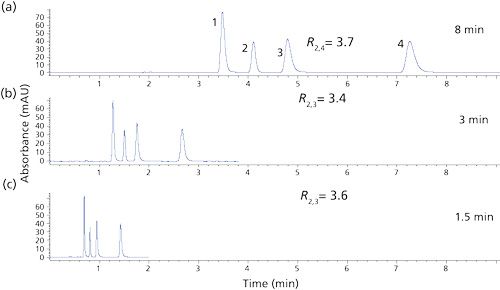
Recently, SPPs under 3 µm have generated significant interest in the chromatographic community (17). A typical example of one of these newer 2.7-µm SPPs is shown in Figure 1c. This configuration has a 1.7-µm solid core with a 0.5-µm shell and 120-Å pores. These particles give rise to column efficiencies rivaling the sub-2-µm TPPs discussed earlier but, because of their 2.7-µm particle size, their pressure drop for the same size column is approximately one half that generated by the sub-2-µm particle. Thus, in addition to UHPLC instruments, most SPP columns of this particle size can be used with some of the conventional 6000-psi (400 bar) rated HPLC systems. Longer columns can be used to generate more theoretical plates with UHPLC systems for more difficult separations. Figure 3 provides a reversed-phase chromatographic comparison of a sub-2-µm column (TPP) with an SPP column run under the same conditions. The chromatograms show almost the same performance for the gradient separation of 17 amino acids. Note that the resolution of the critical pairs (6, 5 and 14, 13) are very similar and the peak widths (w1/2) on both columns are practically the same. However, the pressure is considerably lower on the SPP column. Such performance is typical for modern TPP and SPP columns that use the same surface chemistry.



Newer introductions of SPPs with larger sizes (up to 5 µm) and smaller sizes (down to 1.3 µm) are challenging totally porous columns in the routine marketplace. The smaller particle size SPPs also have challenges since their pressure drops become intolerably high and extracolumn effects of even the most modern commercial instruments can ruin their expected performance. Some newer wider pore SPPs are also available for separations of the larger size biopharmaceutical compounds. A recent 2015 LCGC article by Bell (17) brings us up to date on SPP columns for both large and small molecules, so all of that information that he presented will not be repeated here.
Needless to say, that SPP columns are here to stay and hardly a whisper is heard about sub-2-µm TPPs. In fact, last year’s Pittcon had only a very few TPP columns while SPPs dominated the introductions (18).
Monoliths
Monolith columns do not consist of individual particles but of a continual network of silica or polymer phase that contains two types of pores: flow-through pores with macroporosity (1-2 µm in width) and diffusive pores called mesopores. The mesopores are those that contain bonded phase moieties that control the separation mode. Because of the flow-through pores, the monoliths show very low pressure drops, about half that of a TPP column with the same efficiency and lower than SPP columns of the same dimensions.
It turns out that monoliths have been around for some time; they were introduced several decades ago by BioRad Laboratories as their UNO columns. Never very well-known, these polymeric monoliths are only available with ion-exchange functionality and are recommended for bioseparations. They were never adopted by HPLC advocates. Instead, in the late 1990s, BIA Separations of Ljubljana, Slovenia, produced macroscopic monolithic columns in a disk format and applied them for the separation of biomolecules, namely proteins. With functionalities such as DEAE ion exchanger and protein A affinity and other popular phases, BIA focused their efforts mainly on the preparative and process industry and less so on the analytical size market. Isco, now part of Teledyne, licensed the patents of Cornell University based on the technology developed by Svec and Frechet (19), which was based on rigid macroporous polymer monoliths and began to produce Swift monolithic columns consisting of polystyrene-divinylbenzene functionality. Eventually, Teledyne spun off the product line and sold it to Dionex (now part of Thermo Fisher Scientific) who now markets these columns.
Polymeric monoliths have been studied extensively for the last 20 years or so and at each HPLC conference meeting it leads the pack in column technology presentations with researchers developing new approaches to increase their application potential. Originally, polymeric monoliths were thought to be applicable only to large molecules, but recent work on polymerization techniques has found application to small molecules as well. Unfortunately, very few of these research efforts have led to commercial products that chromatographers can use to solve their separation problems. These polymeric monoliths have the advantage of a wide range of pH compatibility, high loading capacity, and use at high flow rates at low pressures. Yet, only a few products are on the market and only a handful are new.
The first generation silica monoliths were developed by the laboratory of Nakanishi and Tanaka at the Kyoto Institute of Technology in Japan (20). Commercially, they were first introduced by Karin Cabrera of EMD Millipore at HPLC 1998 and introduced at Pittcon by the company as SilicaRod columns in 2000 (21). Later, when new dimensions were developed, the name was changed to Chromolith and a second-generation silica monolith followed later. Unlike polymeric monoliths that can be produced in situ inside the column, silica monoliths are produced in rodlike structures that shrink as the synthesis progresses. Thus, the silica rods have to be cladded into a polymeric sleeve before they could be connected to a pump and detector. In 2001, Merck scientists perfected the technique to tightly clad the silica rod with a PEEK polymer, thereby eliminating well effects and allowing smooth flow down the column matrix. Unfortunately, there were two problems with the technique: Rod lengths were limited to 150 mm and the PEEK enclosure limited the column to a maximum pressure of 125 bar. Thus, to get high efficiency, columns had to be coupled. Since a single monolithic column was already expensive, multiple columns were even worse, compared to conventional packed analytical columns.
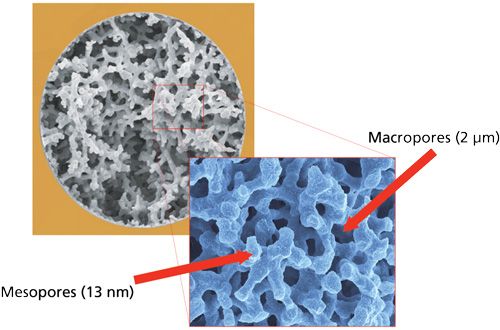
A unique feature of silica-based monolithic HPLC columns over packed particle columns is the ability to independently control the macro- and meso-pore sizes (Figure 4) as well as the silica skeleton size. This control is achieved through the sol-gel preparation process, As a result, this process permits the design of HPLC columns that show simultaneous high separation efficiency and high permeability, not entirely possible with most packed particle columns. In the latter case, the particle size determines the separation efficiency and permeability but in a reciprocal manner-that is, big particles lead to high permeability and low efficiencies whereas small particles lead to the opposite relation. The SPP columns do provide some of the advantages of high efficiency, but with lower permeability than the silica monoliths. By a careful balance of the macro- and meso-pore sizes of the silica monolith, one can balance efficiency and permeability. The macropores allow flow to pass more easily through the column and controls the permeability. The mesopores provide the surface area and control loading capacity and efficiency. As far as chromatographic performance, the initial silica monolith had about the same efficiency as a 3.5-5 µm totally porous particle (22). The big difference between the first-generation (Chromolith) and second-generation (Chromolith HR) columns is the macropore size, which is around 2 µm for the first-generation columns and 1.1 µm for the second-generation columns, respectively. Thus, the Chromolith HR columns are more efficient (140,000 plates/m versus 80,000 plates/m) but have higher pressure drops (65 bar for a 100 mm × 4.6 mm column with a 60:40 (v/v) acetonitrile-water mobile phase flowing at 2 mL/min versus 25 bar for a Chromolith first-generation column) because of the change in the macropore/mesopore domain ratios (22). Still, the newest silica monoliths permit the user to employ conventional 400-bar HPLC instrumentation.


The downside is that this patented technology has not been made widely available since most of the production of silica monoliths is controlled by a single supplier. This low availability has limited the acceptance of silica-based monoliths, especially in the pharmaceutical industry where a second source is often required. In addition, lack of competition and further research and development by other commercial companies has slowed down the potential growth of the silica monoliths.
For those readers who are interested in more details about polymeric and silica-based monolithic columns, Svec published a series of general reviews of these technologies in LCGC starting in 2003 (23-26) in addition to his book on the same subject (27).
Improvements in Other Chromatographic Media
Although silica gel is, by far, the most commonly used LC medium, in the past 25 years a number of other sorbents has been investigated for use in LC columns. Polymers have long been used as chromatographic packings either as pure polymer (for example, polystyrene crossed linked with divinylbenzene [PS-DVB]) or as functionalized PS-DVB (such as sulfonated for ion exchange or derivatized with alkyl chains to resemble reversed-phase media). Other polymers such as polymethacrylate, polyacrylamide, and divinylbenzene also find use as HPLC base materials. Polymers, for the most part, display a much wider pH range than silica gel materials but suffer from a lack of column efficiency and, depending on their degree of crosslinking, may swell or shrink with changes in the mobile phase. Because of their ruggedness, over the years, silica gel-based ion exchangers have given way to polymers where strong buffers, high pH, and higher temperatures are often encountered. Ion chromatography strictly uses polymer-based media. Polymer-based ion-exchange phases with strong- and weak-ionic and ionizable functionalities are widely used in the proteomics and genomics separation area as well as for the separation of other ionic and ionizable compounds, particularly in the area of ion chromatography. Separations of carbohydrates at high pH with pulsed amperometric detection make use of polymeric ion chromatography packed columns.
Since 2003, Pohl has provided constant updates on the improvements of ion-exchange and ion chromatography media (28-32). For more details, readers are referred to his series of excellent articles.
Nonaqueous size-exclusion chromatography (SEC) is also dominated by polymeric media where pore sizes can be easily varied. In recent years, new synthesis techniques have allowed nonaqueous SEC columns to undergo solvent changes without undue swelling and shrinkage. For aqueous SEC, silica gel-based columns do find use. Smaller particle SEC columns have recently become available allowing large molecule characterizations that used to take hours be performed in several minutes. Over a series of articles published in LCGC, Barth and coauthors did an excellent job of keeping readers informed about developments in SEC (33-36).
Other inorganic media that have been introduced over the past 25 years include zirconia, titania, and graphitic carbon, mostly as niche products, but these have never replaced silica as the mainstay in HPLC column packing.
Stationary-Phase Chemistries
The earliest stationary phases used in HPLC with the pellicular packings were coated phases. The packing would be either coated externally and the particles dry-packed or the coatings were applied in situ. The dominant form of liquid chromatography was liquid-liquid chromatography where analytes would partition into the coated phase. Unfortunately, if extreme care was not exercised, the coatings could easily be stripped from the packed bed and retention times drifted. The mobile phase had to be saturated with stationary phase, gradient elution wasn’t feasible, and the column temperature had to be strictly controlled to prevent retention changes. It wasn’t long before researchers developed chemically bonded phases that would negate some of the disadvantages of the coated phases. Early chemical bonded phases of the brush type with Si-O-C bonds were easily hydrolyzed and thus could not tolerate any water or other proton-donating liquids. Siloxane (Si-O-Si-C) bonded phases proved to be more stable and could be used with a wide variety of solvents and pH values. Even today, siloxane phases are the most popular and are available with a wide variety of chemistries.
The availability of bonded siloxane phases had a particularly favorable fallout-the development of reversed-phase LC. The earliest stationary phases were developed for adsorption and normal phase chromatography. These stationary phases were polar (for example, silica gel, Carbowax) and the mobile phase was nonpolar (such as isooctane, hexane). The elution order was nonpolar compounds first followed by compounds of increasing polarity. This was the “normal” practice of HPLC initially. The new mode of reversed-phase LC, never very popular in liquid-liquid chromatography, used the opposite combination of phases from the normal operation. In reversed-phase LC, the stationary phase was nonpolar (such as octadecylsilane, octylsilane) and the mobile phase was polar (for example, water and water-miscible organic solvents like methanol and acetonitrile). Hence, the name (coined by Csaba Horváth) was reversed phase, a name still used today and, by far, the most popular mode of HPLC and UHPLC.
Almost from the beginning of bonded phase packings, reversed-phase LC exploded, especially during the 1970s, and has been widely used ever since. Reversed-phase LC not only can separate nonpolar from polar compounds but can also separate ionic and ionizable compounds by adjustment of the pH via buffers. Many compounds that show low solubility in water and higher solubility in water-miscible organic solvents are amenable to reversed-phase LC; over the years it has even increased in popularity. Not only are popular alkylsilane C8 and C18 phases available but a wide variety of other phases based on cyclic alkyl, aryl, mixed aryl-alkyl, fluorinated, and polar-embedded phases as well as aryl or alkyl ion-exchange mixed-mode phases that have found some niche applications. In a 2010 publication (37), I accounted for more than 1500 reversed-phase columns in over 170 different chemistries that were introduced during the 1970-2010 time frame. More than 92% of all liquid chromatographers use reversed-phase LC at some time in their laboratory (38).
Over the years, improvements have been made in the base silicas used for bonded phases including the development of Type B low trace metal, nonacidic packings providing better separations, and improved peak shapes. Just about every new column introduction made is based on a Type B silica; Type A silicas are now part of history. Exhaustive endcapping was another procedure to remove silanols that cause tailing under certain mobile phase conditions. Silica-organic hybrid phases such as Exterra and bridged-ethyl hybrid (BEH) from Waters (39) offer wider pH capability and can also withstand the high pressures of UHPLC.
Two of the most exciting areas of stationary-phase development over the past 25 years were the advent of chiral phases and phases for hydrophilic-interaction chromatography (HILIC). The former phases were quickly accepted in the pharmaceutical industry since many of the drugs under development have one or more chiral centers and regulatory bodies require the analysis of both enantiomers. In some cases, one of the enantiomers has detrimental side effects for patients while the other enantiomer provides the drug function. One of the highlights in chiral column development was the 2005 introduction of immobilized phases allowing for more rugged columns (40). The introduction of chiral phases for supercritical fluid chromatography (SFC) gave a rebirth to this technique, initially for preparative and process applications, but now for more general purpose chromatography. The introduction of a new generation of SFC instrumentation has spurred on the resurgence. For details on developments in chiral phases in recent years, see the reviews of Beesley (40-45) and Ward and Ward (46,47).
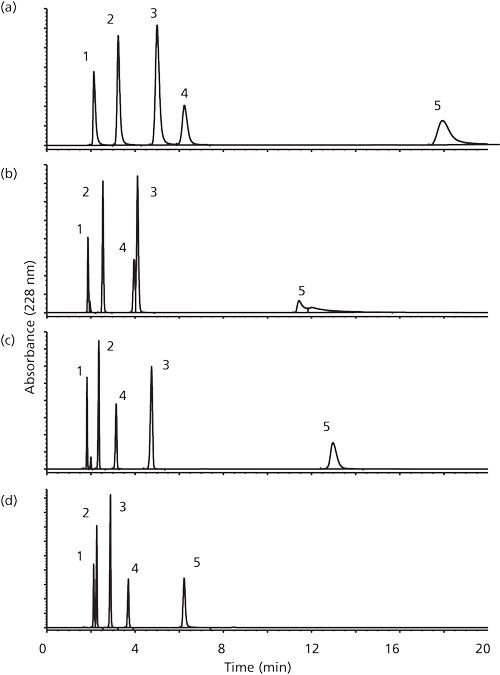
HILIC, which was developed many years ago by Andy Alpert (48) but lay dormant for a number of years, is a separation technique for highly polar analytes that gets around some of the problems associated with reversed-phase chromatography such as low (or no) retention of polar analytes or phase collapse (phase dewetting). HILIC columns often have the opposite effect from reversed-phase LC in that polar analytes generally elute later than nonpolars. Many pharmaceutical compounds contain polar functional groups such as amine and carboxylic acid so HILIC has found widespread use in this marketplace. HILIC uses a polar stationary phase such as bare silica gel, polar bonded phase (such as diol), and certain mixed-mode or zwitterionic phases. Operating conditions usually requires a high percentage of a nonpolar mobile phase, similar to the requirements for normal-phase chromatography. However, unlike normal phase, which uses nonpolar solvents like hexane and methylene chloride and tries to exclude water from the mobile phase, HILIC requires some water (~2%) in the mobile phase to maintain a stagnant enriched water layer on the packing surface into which analytes may selectively partition. In addition, water-miscible organic solvents are used. Under HILIC, polar analytes are well retained and elute in order of increasing hydrophilicity. HILIC is especially favored by MS users since ionization efficiency is often enhanced in organic solvents and the presence of lower concentrations of volatile buffer salt compared to reversed-phase LC. Figure 5, reproduced from the review article by McCalley (49), cites the work of Guo and Gaiki (50) and shows considerable selectivity differences between four silica-based HILIC phases: aminopropyl, amide, zwitterionic ,and bare silica, for the separation of some acidic solutes. They attributed increased retention and different selectivity on the aminopropyl phase to ionic interactions. The improved resolution of aspirin and 4-aminosalicylic acid, compared with that on a bare silica phase was suggested to be caused by electrostatic interaction between the acids and the negatively charged sulfonate groups on the sulfoalkylbetaine phase.


Column Stability and Lifetime
Column lifetime has been steadily increasing over the past 25 years. In our last column survey (51), 25% of the respondents achieved column lifetimes in excess of 18 months and, based on the number of injections, 20% of respondents got at least 1500 injections out of the column. Compare this to 1985 when 53% of respondents had column lifetime of less than 6 months (52). There have been a number of factors that have led to this increase:
- Manufacturers have developed better bonding chemistries for silica gel-based materials with columns now available that can withstand very low pH (<2) as well as those that can reach high pH (pH 12). Phases containing bidentate bonding, organic-inorganic hybrids and sterically protected bonding have extended the pH range beyond the historical 2-8 for siloxane bonded silicas.
- Base materials such as graphitic carbon, zirconia, polymers, and sol-gel silica have greater tolerance to more drastic mobile phase and temperature conditions; however, other problems such as lower efficiency, undesirable surface effects, and so on may have to be tolerated.
- Chromatographers have a much better understanding of the use and limitations of high performance columns and they are more careful in choosing their experimental conditions to maximize lifetime. For example, they don’t wash their silica-based columns with basic solution or run the columns up to their maximum pressure rating.
- Instruments are now more “gentle” on columns not allowing high pressure pulses to slam the column during injection.
- Columns are better packed; manufacturers have developed optimized packing techniques that ensure that the bed doesn’t settle even after extensive use; spherical particles pack more uniformly that the older irregularly shaped particles and thus the beds are more stable.
- Columns are more protected nowadays with in-line filters, guard columns, and better sample preparation to ensure that particulates and undesired impurities don’t get lodged in the column inlet frits or the head of the packed bed.
Expanding on some of these issues, an article that I wrote years ago entitled “The Care and Feeding of Modern HPLC Columns” (53) and another on “Anatomy of an LC Column: From the Beginning to Modern Day” (54,55) are still applicable today.
Specialty HPLC Columns
It was anticipated that, unlike the early days of gas chromatography, a proliferation of LC stationary phases would not occur. Because of the powerful influence of the mobile phase in LC (as opposed to the weak solute-gas phase interactions in gas chromatography), it was thought that mobile-phase optimization would be all that was required to perfect just about any separation. Nevertheless, when a regular column doesn’t do the trick, manufacturers have resorted to tweaked standard phases, new phases with selective chemistries, or even new approaches to achieve desired goals (for example, mixed-mode phases, inclusion complexing, or metal chelation).
Specialty columns are HPLC columns that have been developed for specific separations that are difficult to achieve on a standard column. Sometimes manufacturers will use a standard column, but test it specifically for a certain class of compounds and provide a recommended set of chromatographic conditions. In some cases, the specialty column comes as part of a total solution kit with reagents, standards, and a method. Most specialty columns will be delivered with a test chromatogram from an analysis performed at the factory before shipment and some are guaranteed for a specific separation. Over the years, hundreds of specialty columns have been introduced in my yearly Pittcon articles of new product introductions. Rather than repeating information on each of them, I have chosen to summarize the most popular specialty areas that have appeared in the last 25 Pittcon reports (see Table II).



Other Developments in Columns and Stationary Phases in the Past 25 Years
There have been many other improvements worthy of mention and there is not the space enough to discuss them in detail. So, I have elected to put some of them into Table III, which mostly consists of products that make the life of the chromatographer easier and the explanations while short should convey the improvement that these products have made.


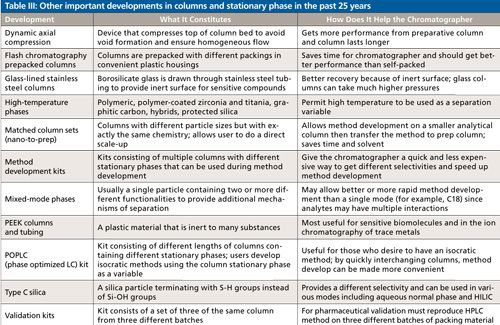
CLICK TABLE TO ENLARGE
Two Hot Topics in the Past 25 Years Involving Extensive Column Technology
There are two topics in which having the right columns and column filling material appropriate for the technique are key elements in the success of the techniques: capillary electrochromatography in the late 1990s to early 2000s and multidimensional and comprehensive liquid chromatography, which is still a hot topic today.
Capillary Electrochromatography
The 1990s was the heyday of capillary electroseparation methods such as capillary zone electrophoresis (CZE), micellar electrokinetic capillary chromatography (MECC), and capillary gel electrophoresis (CGE). Separation scientists were becoming familiar in working with fused-silica capillaries, aqueous buffers, and high voltages. Both practitioners and researchers alike were delving into these electroseparation techniques and instruments were available from the manufacturers. Based on the early work of the laboratories of Pretorius (56), Jorgenson (57), Knox (58,59), and others, using a packed fused-silica capillary was an obvious extension and the technique of capillary electrochromatography (CEC) began to be investigated. A large number of scientists believed in the simple equation of Horváth (CE + C = CEC where CE is capillary electrophoresis and C is HPLC) and that this hybrid technique would eventually take over all liquid phase separations. However, there were fundamental differences between CEC and the other technologies. A nice review on the theory and practice of CEC, published as a featured article in LCGC in 1995 in the early stages of CEC but still current today, was provided by Dittman and coworkers (60).
In CEC, solvent transport is achieved by electroosmotic flow instead of hydraulic flow that occurs in HPLC. The advantage of using electroosmotic flow is that the column efficiency increases because of the plug-flow profile and the ability to use smaller particles than those used in HPLC. Smaller particles can be used in CEC because of the absence of column back pressure when the mobile phase is moved by electroosmotic flow. In the case of CEC, the packed capillary (column) serves as the injector, pump, separation column, and detection cell when detection is performed on-column. The normal chromatographic and electrophoretic parameters that influence retention in CE and HPLC also influence solvent flow rate in CEC since flow is generated by electroosmosis. At the time there was a general agreement that before CEC is an accepted technique, a more fundamental understanding of retention mechanisms must be achieved. But the ability of CEC to combine electrophoretic mobility with partitioning mechanisms was one of its strongest advantages.
In the early days, researchers in the technique stressed its advantages but quickly found out that there were also some major concerns. Poor robustness, low sample capacity, low volume loadability, insufficient sensitivity, lack of commercially available instrumentation, problems with air bubbles, especially around end frits, which halt the CEC experiment, and sample introduction problems plagued the technique at the onset. But probably the biggest single factor that stood in the way of CEC’s acceptance was the lack of that killer application that couldn’t be done by either CE or HPLC alone. Nevertheless, there was much excitement in the chromatography community. At every conference that I went to during those years, there was standing room only with attendees queued in the hallways waiting to get in. However, as the instrument manufacturers didn’t readily invest in CEC and workers continued to face continued experimental problems, interest began to wane. In my notes from HPLC 2001, there were half the number of presentations compared to HPLC 2000. By HPLC 2008, there were only three CEC presentations and none at Pittcon 2008. Perhaps that “killer application” is awaiting someone to come to its rescue with CEC.
Multidimensional and Comprehensive Liquid Chromatography
Multidimensional chromatography has been practiced for many years, both in off-line and on-line modes. Basically, the experiment involves transferring a fraction or fractions from one chromatographic medium (usually a column) to a secondary (or additional) chromatographic medium (column or columns) for further separation. The technique can be used for multiple purposes: for further resolution of complex mixtures that cannot be separated entirely on a single medium, for sample cleanup by removing matrix or interfering compounds, for increased sample throughput, and for trace enrichment of minor compounds of interest. The most popular version of multidimensional chromatography is two-dimensional (2D) chromatography. In the column mode, 2D chromatography has also been referred to as column switching, multiphase chromatography, coupled column chromatography, boxcar chromatography, and sequentialanalysis.
In conventional 2D chromatography, usually one or only a small number of compounds are of interest. One example would be the HPLC separation and determination of a drug in a biological fluid such as urine. The matrix (for example, proteins, uric acids, and so on) is of no interest yet may interfere with the analysis and identification of the drug compound in a one-dimensional (1D) experiment. Other drug metabolites and small molecular weight compounds may also be of zero or little interest. By directing a fraction containing the drug peak plus any possible overlapping contaminants from the primary column to a secondary column, the drug may be measured cleanly provided no other components in the fraction happen to be eluted at the same time on column two.
When the demand is for the complete characterization of complex mixtures, then the term comprehensive chromatography (depicted as LC×LC) is used. Unlike multidimensional chromatography, in comprehensive chromatographic systems every fraction from the primary column is subjected to the second dimension. Normally, fractions are diverted from the primary column (10) to the secondary column (20) at defined time intervals.
To demonstrate an application of LC×LC, Figure 6 presents a pharmaceutical application of LC×LC combined with UV and MS detection. A fully automated multidimensional LC system that could be used for LC-LC (heart cutting) and LC×LC was assembled. This system was used to ensure that there were no coeluted compounds in formulations and to characterize impurities. Coupling normal phase chromatography (diol) as the first dimension using 1-mm i.d. columns with a 100 mm × 4.6 mm C18 monolith column as the second dimension, the authors (61) were able to analyze a fraction every 60 s. Figure 6a shows the separations achieved on the two columns for a proprietary drug mixture. Note the good orthogonal separations achieved on the two different systems. Selectivity was quite different on the two columns. Injection of a tiny volume of immiscible mobile phase containing the fractions of interest onto the reversed-phase LC column was possible without loss of efficiency. Reproducibility was quite good. Finally, Figure 6b provides a colored contour plot resembling a typical 2D profile resulting from the LC×LC and UV coupling experiment.


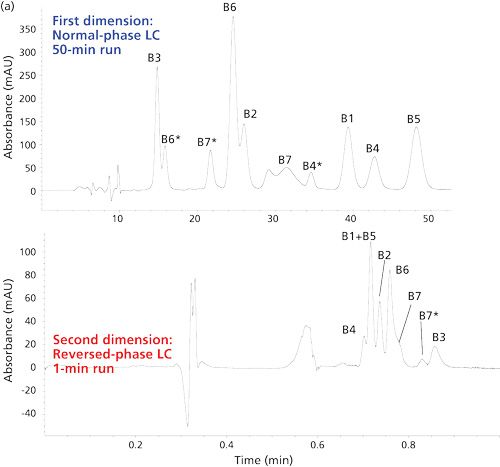



The technique of LC×LC has become quite popular in recent years, made easier by the commercialization of suitable instrumentation. These instruments use alternating trapping columns that allow a certain volume of analyte to be collected (trapped) from column 1, then very rapidly injected onto column 2 via an automatic, high-speed switching valve. The secondary experiment has to be run very quickly and therefore short, fast columns operated at high flow rate is the key to successful coupling. Columns such as monoliths, short SPP columns, and short TPP columns have all found use in this dimension. It is desirable to have the two chromatographically coupled systems be quite different in their chromatographic characteristics, ideally being orthogonal to one another. As difficult samples are encountered, it is expected that multidimensional and the LC×LC technique will be used more and more.
Conclusions
As HPLC enters it golden anniversary in 2016, the past 25 years have seen a continuation of improvements in HPLC and UHPLC columns and stationary phases. Small-particle columns packed into short columns are now the norm with superficially porous particle columns now taking the dominant role. UHPLC is now a household word and applies not only to very high pressure operation, but to the use of these new columns in any mode. Monolithic columns, which have been studied for well over two decades, have not realized their potential, partly because many of the technologies talked about at high-level chromatography symposia do not extrapolate to products that users can actually purchase and use to solve problems in their own laboratories. In addition, only a few suppliers of monoliths are available. The techniques of multidimensional and comprehensive LC dominate almost every chromatography symposium. For complex samples, these techniques offer a greater degree of separation power than can be obtained with a single column and mode.
Silica gel still is the mainstay column material while polymer packings are the first choice in ion exchange or ion chromatography and nonaqueous SEC. Columns for biomolecules (such as proteins and peptides) and for enantiomeric compounds are still in the highest demand. Columns have seen extended lifetimes and workers are now expecting at least 12 months and 1500 injections from a typical column in the reversed-phase mode. The working pH range has been extended for silica-based reversed-phase columns from pH 1.5 to at least 11.5. While reversed-phase LC maintains its overwhelming dominance, HILIC has become the fastest growing LC mode, especially when small polar compounds are encountered.
Interest in CEC, the hottest topic in the late 1990s to early 2000s, subsided when ruggedness and air bubble problems, amongst others, and the absence of suitable commercial instruments, caused workers to go elsewhere. Finding a “killer application” could bring renewed interest in CEC and have it live up to its great potential.
Multidimensional and comprehensive LC×LC chromatography are becoming more popular because of the nature of more complex samples being encountered in HPLC. With instrumentation now available to perform these experiments more easily, these techniques should continue to grow.
References
- R.E. Majors, LCGC12(7), 508-518 (1994).
- R.E. Majors, LCGC North Am. 33(s11), 20-27 (2015).
- L.R. Snyder, J. Chem. Ed.74(1), 37-44 (1997)
- A.J.P. Martin and R.L.M Synge, Biochem. J.35, 1358-1368 (1941).
- E. Piel, Anal.Chem. 38(4), 670-672 (1965).
- P.B. Hamilton, in Advances in Chromatography, J.C. Giddings and R. Keller, Eds. (Marcel Dekker, New York, 1966), vol. 2, p. 3.
- I. Halasz and Cs. Horvath, Anal. Chem.36, 1178 (1966).
- Cs. Horvath, B.A. Preiss, and S.R. Lipsky, Anal. Chem.39, 1422 (1967)
- R.E. Majors, Anal. Chem.44, 1722 (1972).
- R.E. Majors, LCGC21(3), 240-257 (2003).
- B.A. Bidlingmeyer, R.P. Hooker, C.H. Lochmiiller, and L.B. Rogers, Sep. Sci.4, 439-446 (1969).
- B.A. Bidlingmeyer and L.B. Rogers, Anal. Chem.43, 1882-1883 (1971).
- J.E. MacNair, K.C. Lewis, and J.W. Jorgenson, Anal. Chem.69, 983-989 (1997).
- B. Wei, B.J. Rogers, and M.J. Wirth, J. Am. Chem. Soc.134, 10780-10782 (2012).
- R.E. Majors, LCGC North Am.30(s4), 10 (2012).
- J.J. Kirkland, Anal. Chem.64, 1239-1245 (1992).
- D.S. Bell, LCGC North Am. 33(6), 386-395 (2015).
- R.E. Majors, LCGC North Am.32(4) 242-255 (2014).
- F. Svec and J.M.J. Frechet, Anal. Chem.54, 820 (1992).
- N. Tanaka, N. Ishizuka, K. Hosoya, K. Kimata, H. Minakuchi, K. Nakanishi, and N. Soga, Kuromatogurafi14, 50 (1993).
- R.E. Majors, LCGC 18(3) 262-285 (2000).
- K. Cabrera, LCGC North Am.30(s4), 30-35 (2012).
- F. Svec, LCGC North Am.22(s6), 18-21 (2004).
- F. Svec and L. Geiser, LCGC North Am.24(s4), 22-27 (2006).
- F. Svec and J. Krenkova, LCGC North Am.26(s4), 24-30 (2006).
- F. Svec, LCGC North Am.28(s4), 18-23 (2010).
- F. Svec, T.B. Tennikova, and Z. Deyl, Eds., Monolithic Materials: Preparation, Properties, and Applications (Elsevier, Amsterdam, 2005).
- C. Pohl, LCGC Europe16(6a), 51-54 (2003).
- C. Pohl, LCGC North Am.22(s6), 22-25 (2004).
- C. Pohl, LCGC North Am.24(s4), 32-37 (2006).
- C. Pohl, LCGC North Am.28(s4), 24-31 (2010).
- C. Pohl, LCGC North Am. 31(s4b), 16-22 (2013).
- H.G. Barth, LCGC North Am. 22(s6), 43-46 (2004).
- H.G. Barth and G.D. Saunders, LCGC North Am.24(s4), 38-43 (2006).
- H.G. Barth and G.D. Saunders, LCGC North Am.30(s4), 46-53 (2012).
- H.G. Barth and R.E. Majors, LCGC North Am. 31(1), 14-29 (2013).
- R.E. Majors, LCGC North Am. 28(s4), 8-17 (2010).
- R.E. Majors, LCGC North Am.30(1), 20-34 (2012).
- K. Wyndham, T. Walter, P. Iraneta, B. Alden, E. Bouvier, C. Hudalla, N. Lawrence, and D. Walsh, LCGC North Am.30(s4), 20-29 (2012).
- T.E. Beesley, LCGC North Am.24(s4), 28-31 (2006).
- T.E. Beesley and J.T. Lee, LCGC Europe16(6a), 33-36 (2003)
- T.E. Beesley and J.T. Lee, LCGC North Am. 22(s6), 30-33 (2004).
- T.E. Beesley, LCGC North Am.24(s4), 28-31 (2006).
- T.E. Beesley, LCGC North Am.26(s4), 43-46 (2008)
- T.E. Beesley, LCGC North Am. 28(s4), 32-37 (2010).
- T.J. Ward and K.D. Ward, LCGC North Am. 30(s4), 43-45 (2012).
- T.J. Ward and K.D. Ward, LCGC North Am. 32(s4) 20-23 (2014).
- A.J. Alpert, J. Chromatogr. 499, 177-196 (1990).
- D.V. McCalley, LCGC North Am. 26(s4), 53-58 (2008)
- Y. Guo and S. Gaiki, J. Chromatogr. A 1074, 71-80 (2005).
- R.E. Majors, LCGC North Am. 30(1), 20-34 (2012).
- R.E. Majors, LC Magazine 3(9), 774-780 (1985).
- R.E. Majors, LCGC 16(10), 900-909 (1998).
- R.E. Majors, LCGC North Am. 24(8), 740-753 (2006).
- R.E. Majors, LCGC North Am. 33(s11), 33-39 (2015).
- V. Pretorius, B.J. Hopkins, and J.D. Schieke, J. Chromatogr. 99, 23 (1974).
- J.W. Jorgenson and K.D. Lukacs, J. Chromatogr. 218, 209 (1981).
- J.H. Knox and I.H. Grant, Chromatographia 24, 1365 (1987).
- J.H. Knox and I.H. Grant, Chromatographia 32, 317 (1991).
- M. M. Dittman, K. Wienand, F. Bok, and G.P. Rozing, LCGC 13(10), 800-814 (1995).
- Y. Zhao, A. De Villers, P. Sandra, T. Baumgartner, B. Zhang, and J. Kofman, Paper WeL3:3 presented at HPLC 2005, Stockholm, Sweden, 2005.


Ronald E. Majors is the editor of “Column Watch,” an analytical consultant, and a member of LCGC’s editorial advisory board. Direct correspondence about this column to lcgcedit@lcgcmag.com

















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

