Highlights of HPLC 2013, Part II
LCGC North America


Highlights of the column technology observed at the symposium
The 39th International Symposium on High Performance Liquid Phase Separations and Related Techniques (HPLC 2013) was held June 16–20 in Amsterdam, The Netherlands, for the second time. Part I of this coverage reviewed some of the technology and application advances discussed at HPLC 2013 as well as the overall liquid-phase chromatography trends. A summary of the awards presented was also provided. In part II we discuss the column technology highlights observed at the symposium.
The 39th International Symposium on High Performance Liquid Phase Separations and Related Techniques, which alternates between Europe and North America with occasional side meetings in Australasia, was held June 16–20, 2013, in Amsterdam, The Netherlands, for the second time and the third time in Holland. More affectionately known as HPLC 2013, the symposium is the premier scientific event for bringing together the myriad techniques related to separations in liquid and supercritical fluid media. Cochaired by Professors Peter Schoenmakers and Wim Kok of the University of Amsterdam, HPLC 2013 assembled more than 1500 participants from all over the world. This number includes vendor representatives from 55 exhibitors for the three-day instrument, software, and consumables exhibition. The number of conferees was the highest in recent years for this series, with an especially high attendance from European countries, which may indicate that their economic crisis may be abating.
The venue for HPLC 2013 was the colossal RAI building, the location of many Dutch and European exhibits and conferences. The five-day-plus event had a total of 168 oral presentations, perhaps a new record, in plenary and parallel sessions and a whopping 945 posters in sessions with 36 different themes. Lunch was served on-site as part of the registration fee, so conferees didn't have a time-consuming search of restaurants in the area and could focus on science. With an ample social event schedule including three receptions and a symposium dinner and party, 18 vendor workshops, 20 tutorial educational sessions, and four short courses (held during the previous weekend), attendees had their hands full deciding how to allocate their time. The tutorials were particularly well attended and covered current topics such as monoliths, micro- and nanofluidics, hydrophilic interaction liquid chromatography (HILIC), columns and stationary phases, various liquid chromatography–mass spectrometry (LC–MS) topics, applications (environmental, residues, lipidomics, chiral), and comprehensive LC.
This installment of "Column Watch" is part II of my HPLC 2013 coverage. Here, I discuss the column technology highlights observed at the symposium. In part I (1), I presented some scientific highlights of HPLC 2013 and covered the various awards and honorary sessions that took place. Because it was virtually impossible for one person to adequately cover all oral and poster papers, my coverage somewhat reflects a personal bias, although I was able to get presentation notes from some of my colleagues, who are acknowledged at the end of this installment.
New Column Technology Highlights
Many oral, poster, and tutorial sessions were devoted to stationary phases and column technology, always topics of high interest at this series of meetings.
Superficially Porous Shell Particles
From observations made at HPLC 2013, a continuing "hot topic" for the third year in a row was the application of superficially porous particles (SPPs) for developing faster separations and generating more column efficiency at lower pressure drops than the smaller porous particles that they replace. Four years ago, the focus was on comparing sub-2-μm totally porous columns to porous particles in the 2–3 μm range and larger. Three years ago the discussions were focused on the superiority of superficially porous columns over sub-2-μm particle columns. Last year the focus was on the theory and mechanisms on how SPPs work relative to the sub-2 μm packed columns. This year's focus seemed to be mostly on the SPPs themselves. There still are differences in the nomenclature, these particles were referred to as SPPs and fused-core, shell, porous shell, solid core, and poroshell particles. It is now generally accepted that SPPs bring the greatest advantage to users over the smallest porous particles (<2 μm) and, at HPLC 2013, barely a whisper could be heard on the latter technology that spurred the whole topic of UHPLC. The only question now: How soon will both larger and smaller (perhaps down to 1-μm diameter) SPP in various pore sizes and different selectivities be made available? Table I gives a brief update on the current status of commercial SPP products for large and small molecules as of HPLC 2013.


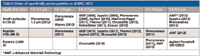
Table I: Status of superficially porous particles as of HPLC 2013
Users who have already purchased UHPLC equipment are less concerned about the lower pressure drops of these columns but for difficult separations, long columns, and lots of theoretical plates are always in demand. Hence, pressure availability is always a concern. Currently, many of the SPP columns on the market do not have the upper pressure limits of some of the UHPLC instruments, but that should change in the near future. However, one aspect of the UHPLC systems that users should be aware of is their greatly decreased extracolumn band dispersion that was optimized for the sub-2-μm columns, especially if interested in columns with smaller internal diameters, such as 2 mm and under.
The majority of the commercial SPPs have particle diameters in the range of 2.6–2.7 μm. Besides the movement of SPPs to larger particle sizes as outlined in last month's article covering J.J. Kirkland's oral presentation (1), Carl Sanchez and colleagues from Phenomenex presented their studies on a 1.3-μm core–shell reversed-phase particles that provide efficiencies approaching 450,000 plates/m (equivalent to ~22,000 plates for a 50 mm × 2.1 mm column). Users feared that frictional heating would be a problem with such small particle columns but core–shell phases provide improved thermal conductivity compared to porous particles of the same dimension so this has not been a factor. In the worst case, for a 50 mm × 2.1 mm column, a radial temperature gradient of 1.5 °C was measured, but in most cases the temperature rise was less than one degree. In a gradient mode, these SPP columns showed a 36% improvement in peak capacity (the best measure of gradient "efficiency") over a 1.7-μm bridged ethyl hybrid column for a bovine serum albumin digest. Although these columns offer some of the highest efficiencies on the market, instruments may not have low enough extracolumn variance to make full use of their power.
The new Waters Cortecs SPP product was introduced at HPLC 2013. The particle diameter is 1.6 μm and the core is 1.1 μm, making the shell 0.25 μm. The plate count for a 50 mm × 2.1 mm column was given as 19,700, about 39% higher than the company's BEH 1.7-μm particle column. The Cortecs column has a higher external porosity (and thus a lower bed density), resulting in a lower back pressure such that the 1.6-μm shell particle column has about the same back pressure as their 1.7-μm porous particle column. It was reported that the new core–shell particle is based on conventional silica rather than the bridged ethyl hybrid chemistry. Similar to other SPP products, the outer surface is rather bumpy. The particle distribution is narrow like other SPPs. The pore size is 90 Å, making it suitable for small molecule separations. Three surface chemistries are provided: C18, C18+ (similar process to charged surface hybrid), and bare silica, with the bonded phases having trifunctional bonding. New low dispersion column hardware is available.
James Treadway of the University of North Carolina in Chapel Hill, North Carolina, reported on the difficulty of translating shell particles to a capillary format. Using 30-, 50-, and 75-μm capillaries with a special carbon fiber microelectrode (no band spreading), for electrochemical detection they packed sub-2-μm experimental shell particles using an acetone-based slurry. The particles were retained using sintered particles as frits. The smallest internal diameter with a 31.7-cm length gave the best performance of 162,000 plates with a reduced plate height (h) of 1.27. For the largest internal diameter, the performance was reduced with h = 1.65 but the particles were slightly different. Other workers have found that packing smaller internal diameter columns with either fused-core or totally porous packings give less efficient columns than for larger internal diameters.
New Column Materials and Related Studies
Each year at this meeting, new column materials are presented in oral research papers, applications papers, poster papers, and at exhibitors' booths. This year was no exception. Tony Edge from Thermo Fisher Scientific presented a paper coauthored with Peter Myers of Liverpool University and others that outlines a totally new particle called a sphere-on-sphere (S-o-S) silica particle (2). Unlike some of the SPP preparation procedures, the S-o-S particles are manufactured using a one-step synthesis delivering a nearly monodisperse particle and core shell morphology. The morphology of the particle has been designed to deliver the real advantages of the core–shell particles. The total diameter of the particle and the core diameter can be controlled together with the effective pore diameters on the surface of the material ranging from 3 to 10 μm with an effective pore size range up to 1.5 μm. The pressure drops observed for packed columns is quite low compared to traditional totally porous particles of similar size. Examples have been shown with bonded-phase S-o-S particles used in normal-phase and reversed-phase separations of small molecules and also proteins. Examples were shown of how these particles can be used as scaffold particles or carrier particles in which new, novel stationary phases can be trapped in the outer layer.
Jeremy Glennon and coworkers of the Innovative Chromatography Group, Irish Separation Science Cluster (ISSC) and Department of Chemistry and the Analytical & Biological Chemistry Research Facility (ABCRF), at the University College Cork, in Ireland, extended their work on core–shell particles incorporating nanoparticles of gold and silica into fused-silica columns and by using capillary electrophoresis enhanced selectivity was shown for low molecular weight biomarkers. With amperometric detection they used a boron-doped diamond electrode that gave greater sensitivity. They also incorporated latex nanoparticles on core–shell and then used them for anion exchange separations on a chip. By coating a fused-silica capillary with gold nanoparticles embedded in a cationic polymer, they were able to separate catecholamine metabolites from urine and aminothiols in plasma. In a related poster paper, they modified a core–shell particle with a chelating functionality (iminodiacetic acid) and used the packing for the high performance chelation ion chromatography (HPCIC) separation of transition and heavy metals. An isocratic separation of a mixture of 14 lanthanides and yttrium required just 8.5 min with one of these core–shell chelators.
Chuck Lucy and colleagues from the University of Alberta talked about carbon phases for ion chromatography (IC) and HILIC. In general, IC gives nice separations of inorganic ions but longer separation times because of the large particle size of the packing materials used. Using small-particle pyrolytic graphitized carbon (Hypercarb, Thermo Fisher Scientific) as a base material, they modified the surface with aromatic carboxylic groups onto the surface via diazonium chemistry and used these modified columns for HILIC applications. These columns appear to have a unique selectivity to commercially available HILIC phases because of the interaction with the underlying carbon. The column is stable in 100 mM sodium hydroxide and provides good separations at high pH unlike silica-based HILIC phases.
Monoliths
Monolith columns have long been desirable because they exhibit high permeability and low pressure drop (because of increased bed porosity), show reasonable separation efficiencies, require no frits to confine the packing material, are easy of fabricate, and nowadays can be made fairly reproducibly. Although this technology has been around for several years, as a routine tool it has yet to see widespread acceptance on the commercial side but improvements continue to be made for both polymer- and silica-based monoliths. Although a number of papers and two excellent tutorials discussed the use of monoliths in various applications, unfortunately, there were few fundamental studies reported on monoliths at HPLC 2013.
Emily Hilder and coworkers from the University of Tasmania in Hobart, Tasmania, Australia gave a keynote lecture on designing polymeric monoliths for chromatography of large and small molecules. Since their introduction more than 20 years ago, polymer monoliths have been shown to provide excellent separation performance for the separation of large biomolecules. Despite incredible promise based on both the ease and flexibility of synthesis, and favorable flow properties, for many people polymer monoliths have failed to live up to the "hype." In particular, the question still remains whether their performance for separations of a wider range of molecules can compete with particle packed columns. Hilder's presentation introduced a wide range of synthesis methodologies and applications that their laboratory has explored to improve both the separation efficiency and selectivity for both small and large molecules using polymer monoliths, demonstrating that these materials can be used for high performance applications. These include grafting phases in layers, incorporating two different functionalities into a single monolith, new synthetic approaches such as the incorporation of nanoparticles into the monolithic structure, or synthesis using cryopolymerization approaches (with and without unidirectional freezing), as well as approaches to extend the operating conditions for these column types, particularly through the use of very high temperature gradients (at temperatures up to 200 °C) or rapid pulses. Separations were shown for both small (for example, inorganic ions and small organic molecules) and large (such as proteins and biopharmaceuticals) molecules. Examples were also provided in which the performance of polymeric monolithic columns was superior to that of particle packed columns.
Although it is a widely used related technique, results obtained using thin layer chromatography (TLC) are not often presented at an HPLC conference. Martin Medal winner Frantisek Svec gave a keynote lecture on monolith columns for TLC separations. In its basic use, this very simple yet powerful separation method does not require any sophisticated instrumentation. His group developed glass-supported monolithic thin layers and used them for the separations of small molecules, peptides, and proteins in both 1D and 2D formats. Svec first shared with the audience several tricks that they were using to prepare well performing layers from a variety of monomers. Whereas poly(butyl methacrylate-co-ethylene dimethacrylate) and poly(glycidyl methacrylate-co-ethylene dimethacrylate) layers with optimized porous structure were prepared using UV-initiated polymerization, poly(styrene-co-divinylbenzene) and poly(4-methylstyrene-co-chloromethylstyrene-co-divinylbenzene) monolithic layers had to be prepared using thermal initiation because these monomers are not UV-transparent. UV-initiated grafting was also used to manipulate the pore surface chemistry. In one of the examples showing excellent performance, they used a moving shutter to spatially control the exposure to UV light, which initiated photografting. This process formed a diagonal gradient of hydrophobic lauryl methacrylate on the top of a hydrophilic monolith. This novel, simple concept was used for the separation of peptides that encountered the gradient during each of the two sequential developments in a 2D regime. In a different experiment, the poly(4-methylstyrene-co-chloromethylstyrene-co-divinylbenzene) layer was hypercrosslinked using Friedel-Crafts alkylation reaction to create a multiplicity of mesopores and significantly increase the surface area. This thin layer then enabled excellent separations of small molecules.
Kazuki Nakanishi and coworkers from Kyoto University and GL Science in Japan reported on recent developments in a macroporous poly(methylsilsesquioxane) (PMSQ) monolith family that has produced a novel type of siloxane-based macroporous polydimethylsiloxane (PDMS) analogs suitable for many kinds of sample preparation purposes. These so-called "marshmallow gels," very soft, low density, highly hydrophobic and highly permeable monoliths with micrometer-range continuous pores, are synthesized by controlled copolymerization of methyltrimethoxysilane and dimethyldimethoxysilane (MTMS-DMDMS) precursors. Because of the presence of dense surface methyl groups, all the surfaces of micrometer-range skeletons are highly hydrophobic exhibiting similar or faster equilibration rate than those of other PDMS-based solid-phase extraction (SPE) devices. Physical properties are comparable to conventional PDMS materials; marshmallow gels exhibit no degradation under temperatures as high as 600 K, and no obvious glass transition in the temperature range as low as 150 K. Furthermore, even under liquid nitrogen (77 K) the material still exhibits rubbery elastic deformation and recovery. In the above broad temperature range, the extraction of hydrophobic substances from a polar solvent system can be dramatically accelerated by absorbing the mixture and simply squeezing it out. The marshmallow gels may find use in both in GC and LC sample preparation purposes. Not limited to the PDMS analog, the chemical modification of the pore surface is easy by choosing appropriate precursors. Typical processing requires only a few hours in any shape and size below 100 °C.
HILIC
HILIC is still one of the most popular chromatographic techniques, particularly for small polar analytes that are eluted from reversed-phase columns way too fast. One of the most interesting papers studying the mechanism of HILIC separations was presented by Ulrich Tallarek of Philipps University in Marburg, Germany. He modeled the entire HILIC operation. It is generally agreed that the mechanism of HILIC retention involves the water-rich layer at the polar stationary phase boundary. He contends that retention in HILIC shows a shallow increase with increasing acetonitrile, but then a sudden leap in retention occurs at 75–95% acetonitrile. Water and acetonitrile prefer to be in an environment of their own kind (like prefers like). He postulated that there are three distinct regions in the pores:
- Immediate surface region: The rigid layer; here the silanols strongly adsorb water. This region is "ice-like" where the water molecules have a long residence time that may be 100 times larger than in the bulk region. The composition of water is unchanged no matter what the concentration of water is in the mobile phase. Here, the water molecules are all bound to the silanols.
- Intermediate (interface) region: The diffuse layer; this region responds somewhat to the water concentration in the mobile phase but lags behind it. Here it quickly decays to water-water hydrogen bonds. It gradually relaxes to the bulk bonded phase.
- Bulk region: In the bulk region, there is some hydrogen bonding between water and the acetonitrile.
Even at 95% acetonitrile, the water molecules are mostly in contact with other water molecules. Methanol is much more like water so it behaves quite differently in HILIC compared to acetonitrile. The ratio of water mole fraction at or near the surface and the bulk water concentration grows nonlinearly with the acetonitrile fraction in the mobile phases and ends with a "jump" thereby explaining the sudden leap in retention at higher acetonitrile concentrations. Regions 1 and 2 make up about 56% of the pore volume.
Column Hardware Design
Every once in a while a new column hardware design comes along that proves quite interesting. This year's HPLC meeting saw an innovative concept introduced by Andrew Shalliker of the University of Western Sydney in Australia. His talk was entitled "Active Flow Technology Presenting the Infinite Diameter Column in a Curtain Flow Mode of Operation." Many years ago, Professor John Knox noted that an infinite-diameter column would be the best type of column of all because the analyte injected into the center of a column would never reach the walls where the flow velocity profile is upset by the particle–wall interface. So for many years injectors were designed to inject the sample into the center of the column. Most likely though, the analytes did diffuse in a radial direction and did reach the wall to disrupt the nonparabolic flow that one would like. Active flow technology consists of two types of column designs: curtain flow technology refers to the process of injection of sample across a radial cross section of the column to ensure the analyte sees just the middle of the packed bed where the flow is not disturbed by wall effects. Parallel segments flow relies on sampling the column effluent in the very middle of the column to also ensure the absence of wall effects, which again gives the best overall column efficiency. How is this done? Figure 1 is a special column hardware design used to sample flow near the walls as well as in the middle of the elution profile so that the parabolic flow pattern could be sampled. By segmenting the flow and selecting just the middle portion, improved efficiency was obtained by this virtual "wall-less" column.

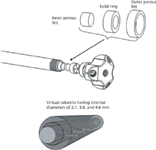
Figure 1: Design of an active flow technology column. Courtesy of Andrew Shalliker of the University Of Western Sydney in North Parramatta, Australia.
If you have 21% of the total flow on a 4.6-mm id column passing through its center, you emulate a 2.1-mm virtual column. If you have 43% of the total flow going through the center then you emulate a 3.0-mm virtual column. These observations are the same as cutting the parabolic Poiseuille flow profile, especially evident at a high flow rate. Shalliker first showed data for a regular monolithic column with normal endfittings with 107,116 total plates with a tailing factor of 1.5. With the curtain flow technology, the same column resulted in a total plate count of 111,813 and a tailing factor of 1.14, a definite improvement especially with the latter. He also indicated that a column packed with SPPs behaved better than a column packed with totally porous particles.
Extracolumn Contributions for Modern High Efficiency Columns
Extracolumn contributions to band spreading has also become an important topic with the high efficiency SPP and totally porous columns now commonplace. Extracolumn contributions are defined as any volume outside of the column bed itself from the point of sample introduction to the exit of the detector and includes the injector, connection capillaries, endfittings including frits, and the detector cell volume. Most older instruments do not have the ability to effectively work with these modern columns and if such a column is installed into the chromatograph, it will have poor performance. When column volumes decrease, such as what is encountered when moving to short 2.1- or even 1.0-mm id columns, the problem becomes magnified. Adding to this topic of extracolumn dispersion effects highlighted in last month's coverage of the Horváth Award lecture (1) was a keynote lecture by Monika Dittmann of Agilent Technologies, where she showed the practical limitations of further decreases in extracolumn effects such as the high pressure generated by small-internal-diameter capillary tubing (for example, ~50-μm i.d.). For low k' (such as <3) analytes on 2.1- and 1.0-mm i.d. columns, the extracolumn variance has a larger impact in isocratic separation than in gradient work, and for a 50 mm × 2.1 mm column with modern superficially porous or sub-2-μm totally porous particles, the variance must be reduced to below 1 μL2 to realize the full potential of today's high efficiency columns. For gradient separations, the external contributions have a smaller influence because of the larger peak volumes and the focusing of the analytes at the head of the column. For UV detection, she showed that 250-nL flow cells may be preferred over 800-nL flow cells for fast gradients and low k' isocratic analysis. Fixed-loop injectors may not be the best for low dispersion and newer autosampler needle seat designs with a low variance contribution (~1.5 μL2) may be the answer. When using electrospray ionization (ESI)-MS peak capacities are comparable to optimized UV detection, but when low sampling rates are used in MS, the peak capacity can be lower for fast gradients in high efficiency columns.
Gert Desmet of the Free University of Brussels in Belgium presented a tutorial on system performance. He presented an overview of the underlying theory needed to understand the influence of the instrument design (system and dwell volume, oven design and operating mode, flow rate, and pressure range of the pump) on the observed column performance. A useful part of his lecture showed the audience how to calculate the extracolumn variances and other practical parameters of the various parts of HPLC and UHPLC systems. These calculations may be of importance because of the difficulties of measuring these quantities experimentally. Some useful equations were presented. For example, to calculate the pressure (ΔP) imposed by empty capillary tubing, equation 1 was presented:

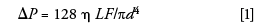
where η is the mobile-phase viscosity, L the column length, F is the flow rate, and d the internal diameter.
Pressure Effects in HPLC and UHPLC
Since the advent of UHPLC and with pressures sometimes greatly exceeding the 400-bar limits of yesteryear, the impact of column pressure on retention has brought about concerns that were never expressed before. We briefly addressed the myth "pressure has no effect on retention time" in a recent article (3). This year at HPLC 2013 two notables in the field, David McCalley of the University of the West of England in Bristol, United Kingdom, and Nobuo Tanaka, now with GL Science in Japan, presented papers on pressure effects in UHPLC.
McCalley evaluated retention time changes with different pressures for five different 5-μm stationary phases, all with the same mobile phase, using a restriction capillary at the end of his system. He confirmed Martin and Guiochon's results on the effects of the compressibility of the organic portion of the mobile phase. He also showed that almost all solutes showed the same effects. As the pressure increased from 53 to 246 bar there were some retention time changes resulting in some peak elution reversals that he interpreted as coming from selectivity changes, although efficiency (N) was not affected. Comparing "rigid" to "flexible" molecules, there are more changes in retention time for the more rigid (planar) molecules with pressure increases. When comparing monomeric and polymeric stationary phases, different effects are seen, with the polymeric stationary phase showing more changes. Within this category, polar and ionizable compounds show larger effects than nonpolar ones, although size also has an influence.
Tanaka investigated pressure effects from 7.6 to 48 MPa on the separation of monounsaturated fatty acids, using a polymeric stationary phase. Peak symmetry is better at higher pressures. A similar effect was seen with fatty acid methyl esters (FAMEs). Comparing a monomeric C18 stationary phase with a C30 stationary phase, he reported that the C30 showed reasonable separations for some FAME and some triacylglycerols while the C18 phase was not selective. On the other hand, the monomeric C18 could effect separation of the α, β, and γ tocopherols, although polymeric C18 was better. The elution order is different for the tocopherols on C18 versus C30. One interesting observation was that lower temperatures improved separation on the C30 phase by increasing retention time and that combining lower temperature with increased pressure gives even better results.
Tanaka suggested three categories of pressure effects on compounds:
- Neutral nonpolars — small effects
- Weak acids and bases near their pKa — changes of pKa with pressure can cause changes in retention
- Ions, neutral hydrophilic compounds
The outcome of his lecture seemed to be that certain selective stationary phases can give interesting changes in selectivity with pressure. Increasing the pressure can increase the retention of a favored isomer. The C18 and C30 stationary phases gave a big difference for flexible molecules. If there is a closer distance between the solute and stationary phase caused by pressure increases, dispersive interactions will increase. Nonselective stationary phases will not show these differences.
Multidimensional and Comprehensive Liquid-Phase Separations
This year multidimensional separations were a hot topic. In the 2D short course, presented the weekend before HPLC 2013, more than 50 conferees elected to come early to Amsterdam to learn about the latest technology. One of the better educational-themed presentations was that of Paola Dugo of the University of Messina in Messina, Italy, whose tutorial "Comprehensive 2D LC" pointed out some of the problems encountered including
- Coupling of different modes is difficult.
- Problems are encountered with immiscible solvents in the two modes.
- Specific software is needed.
She amply pointed out that in heart-cutting chromatography equation 2 holds:

where Nc is the total peak capacity of the combined LC columns, c1 and c2 whose individual peak capacities are Nc1 and Nc2, respectively, while in comprehensive LC (usually depicted as LC×LC), equation 3 holds:


which shows the strong separation power of comprehensive chromatography.
To obtain high 2D resolution, each peak in the first dimension should be sampled at least 3–4 times to prevent the loss of 1D information. You have to think that if you have a 1D peak with three components and three different retention times, then each component will show in 2D, but to different extents.
To connect the two columns, a 10-port valve with two loops can be used. One can use two sampling loops although two trapping columns may also be used. In the former case, the loops must be large enough to hold the entire volume content from the effluent of column 1 while the complete separation of the contents of the other loop is being injected onto and separated on column 2. Generally, in the final setup, one detector is needed because all the material from 1D is transferred to 2D; thus there is no need for a 1D detector. Generally, the 1D column is long and operated at low flow. The 2D column must provide fast analysis, usually a couple of minutes or less. Here, a narrow-bore column can be used to prevent sensitivity loss. Fast gradients are very common. The column needs to have a fast equilibration time so core–shell, monoliths, or sub-3-μm columns are frequently used. It is possible to use an array of 2D columns in parallel. The problem with that approach is that two columns are rarely identical. However, if you use two columns in the 2D, you can half the 2D analysis time because the one 2D column can be reconditioned while the first 2D column is performing the analysis. Usually the modulation time is 30 s to 2 min.
Dugo also showed some nice food applications examples in her presentation including lipid analysis (such as triacylglycerols and phospholipids) and a combination of reversed-phase (separation based on hydrophobicity) and argentation (Ag+) chromatography (separation based on degree of unsaturation).
Kelly Zhang of Genentech talked about 2D LC applications in pharmaceutical chemistry. Some of her practical applications included excipient analysis (for example, mannitol and lactose) by HILIC, salt in active pharmaceutical ingredients (APIs) using multimodal columns, a stability indicating method, quantification of coeluted impurities, separation of multiple chiral center compounds, and 2D LC–MS switching from a non-MS-compatible mobile phase to an MS-compatible mobile phase.
Thorsten Teutenberg and coworkers from the Institut für Energie- und Umwelttechnik e. V., Duisburg in Duisburg, Germany, strongly supports the use of LC–LC (selective fraction collection) or LC×LC (continuous fraction collection) to reduce dependence on the "magic" of MS, especially for environmental samples. The use of nanoLC in both dimensions helps reduce the mobile phase "incompatibility" problem. His application example was wastewater in which he encountered >60 compounds.
HPLC 2013 (Australia) and HPLC 2014 Are Next
The next major symposium in this series, the 40th Symposium, will be November 18–21, 2013, in Hobart, Tasmania, Australia. The chairman of this meeting will be Paul Haddad, who will be retiring after HPLC 2013 Australia. For more information go to www.hplc2013-hobart.org. In May 2014, the series returns to the United States for the 41st Sympoisum. HPLC 2014 will be held in New Orleans, Louisiana, always a favorite place to visit. The Symposium Chair will be J. Michael Ramsey of the University of North Carolina in Chapel Hill, North Carolina. You can learn about this meeting at the following web site: www.hplc2014.org. Bookmark both of these web sites so that you can keep up on the latest happenings.
Acknowledgments
I would like to acknowledge the contributions of my Agilent colleagues, who supplied notes on some of the sessions: Xiaoli Wang, Wu Chen, and Norwin Von-Doehren. A very special thanks goes to Maureen Joseph, also of Agilent, who took very copious and thorough notes at many of the columns sessions. Also, I would like to thank Professors Carol Collins from the University of Campinas in Brazil and David McCalley of the University of the West of England for sharing their notes with me.
References
(1) R.E. Majors, LCGC North Am. 31(9), 770–776 (2013).
(2) A. Ahmed, W. Abdelmagid, H. Ritchie, P. Myers, and H. Zhang, J. Chromatogr. A 1270, 194–203 (2012).
(3) R.E. Majors, LCGC North Am. 31(7), 522–537 (2013).
Ronald E. Majors "Column Watch" Editor Ronald E. Majors is a Senior Scientist in the Columns and Supplies Division at Agilent Technologies (Wilmington, Delaware), and is a member of LCGC's editorial advisory board. Direct correspondence about this column to lcgcedit@lcgcmag.com.


Ronald E. Majors













Perspectives in Hydrophobic Interaction Temperature- Responsive Liquid Chromatography (TRLC)
TRLC can obtain separations similar to those of reversed-phase LC while using only water as the mobile phase.



