Frontiers in Ultrafast Chiral Chromatography
LCGC North America
We explore the developments that have made it possible to produce sub-minute, and, more recently, even sub-second enantiomeric separations. We also look at the questions that remain unanswered.
Compared to the progress made in reversed-phase separations in terms of speed and efficiency, chiral chromatographers have traditionally focused on improving the selectivity of enantiomeric separations by synthetic procedures. As a result, more than 50 different types of advanced chiral stationary phase chemistries are available today. Traditionally, speed and efficiency of chiral chromatography has not received attention until recently. With the availability of superficially porous particles and sub-2-µm particles, sub-minute enantiomeric separations have been achieved with the help of improved particle technology with narrow size distribution, and systematic studies on packing columns. This article covers advances made in the field of ultrafast chiral chromatography in the last decade. The development of instrumentation has also contributed immensely to making sub-second chiral separations a reality. Enantiomeric separations can now compete with the speed of sensors. Future directions and unanswered questions in the field of ultrafast enantiomeric separations are highlighted.
Ultrafast chiral chromatography optimization aspires to have a resolution (Rs) of 1.5 between a pair of enantiomers in the shortest possible time. Advances in stationary phases and instrument technology have changed the magnitude of the shortest possible time from nearly an hour to under a second. In the 1960s, most separations were commonly performed within the time frame of an hour (1). However, around this time, some researchers were reducing analysis times in achiral liquid chromatography (LC) by increasing the flow rate, using shorter columns packed with smaller particles, and using narrow diameter columns to increase the linear velocity (2). In high performance liquid chromatography (HPLC), a separation of three components in under 3 min was achieved on silica gel in the normal-phase mode as early as 1971 (2). In gas chromatography (GC), a sub-minute separation of five components was performed with open capillaries in the late 1980s (3). Though sub-minute separations using supercritical fluid chromatography (SFC) were known to chiral chromatographers since the early 1990s (4), ultrafast separation of enantiomers using LC is currently considered as one of the most challenging types of separation (5).
Chromatographic theory indicates that the resolving power of any column depends on selectivity, efficiency, and retention factor. Decades of research involving new chiral selectors produced columns with large selectivities (α), such as cellulose or amylose derivatives, macrocyclic glycopeptides, cyclodextrins, cyclofructans, and π-complex phases on 5-µm particle silica supports. Historically, most enantiomeric separations had poor plate counts; for example, 40,000 plates/m in 25-cm long columns. This has changed in recent times, and enantiomeric separation efficiencies are now approaching achiral separation efficiencies. The sudden impetus for ultrafast chiral chromatography is a result of multiple advances. The use of superficially porous particles (SPPs) and sub-2-µm fully porous particles (FPPs) is an important factor in bringing chiral separation efficiencies on par with C18 columns. In addition, state of the art column packing methods has led to a rise in efficiency for enantiomeric separations from 50,000 plates/m to 250,000–300,000 plates/m (6,7). The instrumental advances have significantly contributed to ultrafast chiral chromatography as well, particularly in terms of reducing extracolumn dispersion, higher flow rates, and better detectors. Figure 1 illustrates how enantiomeric separations have evolved over the years with respect to analysis time from 5–30 min to sub-minute analyses (8). This article will discuss the breakthroughs in ultrafast enantiomeric separations, and will conclude with the latest developments in the field that have managed to achieve separations in the sub-second domain.


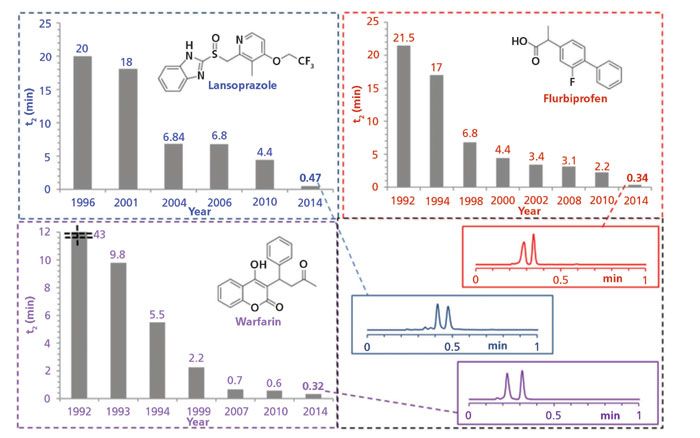
Figure 1: Historical evolution of speeds for chromatographic separation of the enantiomers of lansoprazole, flurbiprofen, and warfarin. Adapted with permission from reference 8.
The Particle Debate in Ultrafast Chiral Chromatography
As chiral stationary phase supports, fully porous particles (FPPs) and superficially porous particles (SPPs) have been debated in the literature as chromatographers have tried to single out a particle morphology best suited for ultrafast separations (6,7). As is evident from the van Deemter equation, smaller particles can offer lower theoretical plate heights (9),


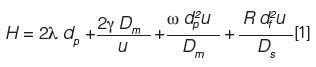
where dp = particle diameter, H = height equivalent to a theoretical plate, Ds = diffusion coefficient of the stationary phase, df = film thickness, dc = capillary diameter, Dm = diffusion coefficient of the mobile phase, λ = particle shape, and γ, ω, and R are constants. Though all the terms in the equation contribute to the broadening of an analyte band, attention to the particle size term in the equation is made herein. Smaller particles result in a lower contribution to band broadening from mass transfer, along with a significant decrease in eddy dispersion. The decrease in particle size appears to be the obvious way forwards for chiral chromatographic columns, but the resulting increase in column back pressure somewhat mitigates the advantages of smaller particles.
The high efficiencies of the sub-2-µm particles are of paramount importance when performing ultrafast chiral chromatography. However, a paradigm shift in particle technology was not possible without overcoming certain hurdles. First, during the synthesis of the stationary phase, smaller particles tend to aggregate more readily than larger particles. Aggregation of smaller particles in poorly dispersive solvents can result in low and nonreproducible surface-bonded chemistry (10). Furthermore, column permeability decreases with the reduction in particle diameter, as described by the Kozeny-Carman relation, which shows that permeability is inversely proportional to (dp)2 (11). For example, an approximate ninefold increase in back pressure occurs when the particle size is decreased from 5 µm to 1.7 µm. Large particle size distribution also creates problems. Tremendous advancements in the field of ultrahigh-pressure liquid chromatography (UHPLC) instrumentation have made the use of these smaller particles possible for enantiomeric separations.
Fully Porous Particle Chiral Stationary Phases
Gasparrini and co-workers were the first to bond a chiral selector to sub-2-µm FPP particles to perform ultrafast enantiomeric separations (6,12). Upon theoretical investigation, their van Deemter plots revealed a flat C-term profile up to 14 mm/s. High efficiencies were observed at higher than optimal flow rates as encountered in routine HPLC. A wide array of ultrafast chiral separations were shown. Gasparrini described the process of transitioning from conventional 5-µm particle-based enantiomeric separations to sub-2-µm particle-based enantiomeric separations with UHPLC. The intrinsic kinetic performances of sub-2-µm silica were retained after bonding the chiral selector to the particles. An ultrafast separation using sub-2-µm particles at multiple flow rates is shown in Figure 2.


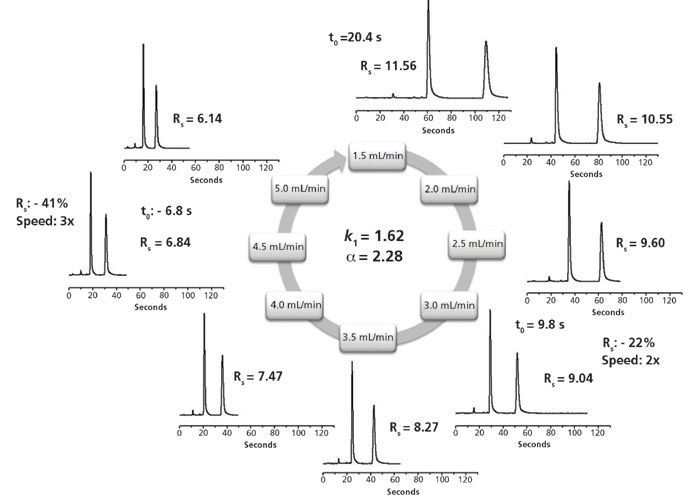
Figure 2: Ultrafast resolution in polar organic mode (POM) of the enantiomers of naproxen on the UHPLC 50 mm × 4.6 mm, 1.7-µm (R,R)-Whelk-O1 column at flow rates (mobile phase: acetonitrile + 0.2% acetic acid + 0.07% DEA (v/v/v), UV detection at 254 nm, Tcol = 25 °C). Adapted with permission from reference 6.
In the 1970s, the efficiencies of large particle-based columns were stated to remain unaffected by the size distribution of the particles as long as the deviation was lower than 40% (13). In contrast, Desmet and co-workers recently reported a linear dependence of reduced plate heights with narrow particle size distribution (14). In agreement with Desmet, Catani and colleagues showed experimentally higher efficiencies for columns packed with narrow size distribution particles compared to polydisperse particles (15). The first narrow size distribution particles were 1.9-µm FPPs. Size distribution for these particles has a standard deviation as low as 6% (16). Barhate and colleagues bonded macrocyclic glycopeptides (teicoplanin, vancomycin, and teicoplanin aglycone) to narrow size distribution silica particles (16). They reported efficiencies up to 210,000 plates/m. Lower reduced plate heights were reported with columns packed with narrow size distribution particles compared to polydispersed 1.7-µm particles. Different classes of chiral compounds, namely chiral heterocycles, amino acids, β-blockers, and pharmaceutically important drugs, were enantiomerically separated with UHPLC and SFC. Ismail and colleagues reported similar observation with the narrow size distribution silica particles, and used them for ultrafast separations (17). A problem with these small particles is the resulting frictional heating, which had been predicted four decades ago by Halász in his classical paper "Ultimate Limits in High-Performance Liquid Chromatography" (13), and will be discussed in a later section.
Superficially Porous Particles Bonded Chiral Stationary Phase
As pointed out earlier, sub-2-µm particles generate higher back pressures as a result of reduced column permeability. Such particles cannot be used in common HPLC systems because of limitations regarding the pressure that can be tolerated. Specially designed pumps for UHPLC systems are required. Hence the search for a particle morphology that can provide efficiencies comparable to that of sub-2-µm fully porous particles, but with lower back pressures, led to the advent and use of 2.7-µm SPPs for chromatographic columns. SPPs, or core–shell silica particles, are chromatographic supports that possess a solid, impenetrable core. This particle morphology has been applied to achiral columns and has led to high efficiencies (reduced plate heights 1.4–1.6). The SPPs exhibit much better packing homogeneity compared to FPPs (18). Better packing homogeneity across the column radius has led to a lower contribution to band broadening, owing to lower eddy dispersion contributions (18). SPPs are also able to decrease other factors that contribute to band broadening, such as longitudinal diffusion and resistance to mass transfer.
Initial reports in 2012 described coated polysaccharide chiral selector on SPPs as showing decreased enantiomeric selectivity compared to its fully porous counterpart (19). A reason for this observation was attributed to the reduced chiral selector loading and difficulty in reproducing coated phases on SPPs owing to their small pore size (~100–400 Å) compared to wide pore FPPS (pore sizes in the range of 1000 Å), which are used to manufacture coated chiral stationary phases.
A significant advance in the field of SPP-based chiral stationary phases occurred when bonded brush-type cyclofructan-based and macrocyclic glycopeptide chiral selectors were used. Much higher efficiencies were obtained than on FPPs, while retaining or improving enantiomeric selectivity under the same mobile phase conditions (20). Another important take-away from the study pointed to the higher efficiency and significantly lower analysis times of the SPPs compared to the FPPs across different chromatographic modes, such as reversed phase, polar organic, and normal phase.
The subsequent systematic study conducted by Armstrong and colleagues highlighted the advantages of SPPs for manufacturing chiral stationary phases, and performed ultrafast separations using 2.7-µm SPPs (7). The authors reported comparisons of enantiomeric separations on columns with multiple particle diameters and different particle morphologies ranging from 5-µm to 2.1-µm FPP and 2.7-µm SPP-based chiral stationary phases, some of them commercially obtained and some produced in-house. The superior performance of the SPPs to the commercially available chiral columns was quite apparent. Reporting reduced plate heights as low as 1.6, the group performed chiral separations for a broad range of an important class of molecules (all under a min), using different chiral selectors, namely teicoplanin, teicoplanin aglycone, vancomycin, hydroxypropyl β-cyclodextrin, and derivatized cyclofructans (Figure 3).


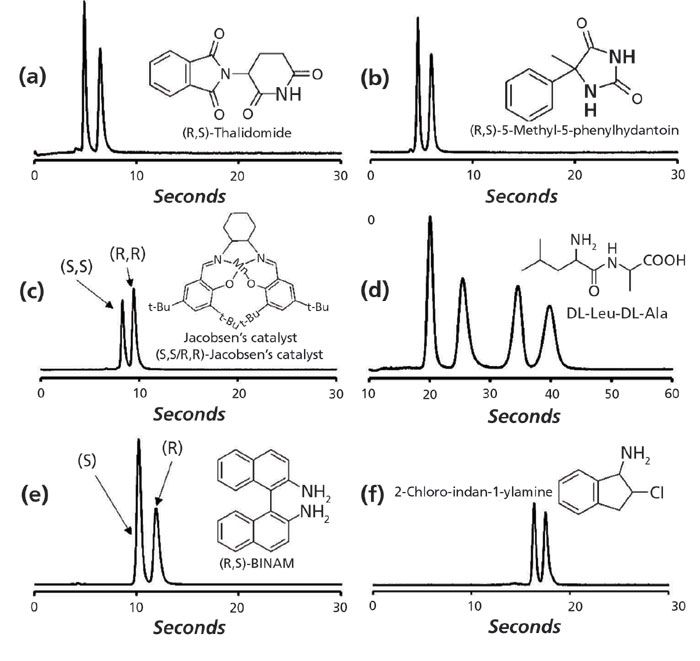
Figure 3: Representative ultrafast enantiomeric separations on each of six chiral stationary phases: (a) vancomycin SPP (3 cm × 0.46 cm, 2.7-µm), MP = methanol, 4.95 mL/min, Tcol = 60 °C, (b) teicoplanin aglycone SPP (3 cm × 0.46 cm, 2.7-µm), MP = methanol, 4.70 mL/min, Tcol = 60 °C, (c) hydroxylpropyl-β-cyclodextrin SPP (5 cm × 0.46 cm, 2.7-µm), MP = 97:3:0.3:0.2 acetonitrile-methanol-TFA-TEA, 4.75 mL/min, Tcol = 60 °C, (d) teicoplanin SPP (3 cm × 0.46 cm, 2.7-µm), MP = 40:60 water-methanol, 3.00 mL/min, Tcol = 22 °C, (e) CF7-DMP SPP (3 cm × 0.46 cm, 2.7-µm), MP = 90:10 heptane-ethanol, 4.80 mL/min, Tcol = 22 °C, (f) CF6-P SPP (10 cm × 0.46 cm, 2.7-µm), MP = 70:30:0.3:0.2 acetonitrile-methanol-TFA-TEA, 4.50 mL/min, Tcol = 22 °C. All columns were manufactured by AZYP LLC. Adapted with permission from reference 7.
The analysis time was further reduced by Wahab and colleagues, who performed sub-second enantiomeric separations of multiple biologically important molecules (21). The authors successfully packed 0.5-cm columns of variable internal diameter (0.46 cm, 0.30 cm, and 0.21 cm) and reported enantiomeric separations at unprecedented speeds. Their work pointed out the necessities of multiple hardware improvements. Other reports have also pointed out the superiority of 2.7-µm SPPs over traditional 5-µm particles using geometry independent kinetic plots (22). The 5-µm particles investigated lagged both in efficiency and analysis times when compared to the 2.7-µm SPPs. The geometry independence of their results highlights the considerable impact of particle morphology on column efficiencies.
Min and colleagues tried to incorporate the advantages of both SPPs and sub-2-µm particles and synthesized 1.5-µm teicoplanin-bonded SPPs to demonstrate the potential of sub-2-µm SPPs (23). This is the only report available in this area that the authors are aware of and further studies will hopefully be conducted in the future.
Do we have an answer to the question of best particle morphology? No. A 2016 study by scientists at the University of Roma found, despite the previously stated advantages of SPPs, that columns with sub-2-µm FPPs outperformed the 2.7-µm SPPs. Furthermore, a study conducted by Ismail and colleagues highlighted the superior performance of 1.8-µm and 2.5-µm FPP compared to the 2.6-µm SPPs. They attributed the larger surface density of the chiral selector on the SPPs as a probable factor for the slower mass transfer kinetics leading to lower efficiencies (24). Recent work by the same authors using teicoplanin bonded to 2-µm SPPs has produced excellent results with efficiencies reaching 300,000 plates/m (25). This study compared the SPPs with sub-2-µm narrow size distribution silica particles, and found the 2-µm SPPs to be superior in terms of efficiency and more promising toward the development of ultrafast chiral chromatography than the 1.9-µm FPPs. The findings mentioned above by independent research groups will hopefully help SPPs gain traction in the field of chiral separations. Nonetheless, particle morphology is still a hotly debated topic both from a theoretical and experimental perspective, and further research is needed to reach a consensus.
However, the major shift in particle size of the silica support from 5- to sub-2-µm has led to its adoption in multiple applications, such as ultrafast and two dimensional chromatography. The commercial availability of these columns has also helped smaller particles gain popularity. Both FPPs and SPPs have recently been used to perform multiple separations in two-dimensional liquid chromatography (2D-LC), and the promising results are sure to pave the way for further development in this field. In the past, enantiomeric separations were rarely performed in the second dimension of 2D-LC, because of the slower mass transfer kinetics and problems with peak wrap-around, but recent advances in speed have allowed for the use of such chiral stationary phases in the second dimension (26).
Frictional Heating Issues with Ultrafast Chromatography
Frictional heating is an inevitable consequence of high flow rates in ultrafast achiral or chiral chromatography. Frictional heating has been documented by researchers by measuring the flow averaged temperature at the column outlet with a 10 °C rise in mobile phase temperatures found (7). Though frictional heating has been found to cause distortions in peak shapes in conventional reversed-phase chromatography, it can be beneficial when performing ultrafast chiral chromatography (7). It is well known that frictional heating can be broadly classified into two types: radial and axial. Scientists studying the effects of frictional heating on chiral separations found that axial temperature gradients improved peak efficiency, but radial heating was detrimental to efficiency (7). A radial temperature gradient lowers column efficiency as a result of the distortion of the laminar flow profile. From mathematical treatments of radial heating, it can be concluded that high flow rates, higher pressure drops across the column, large column radius, and poor thermal conductivity of the mobile phase increase temperature gradients in the radial direction (center being the hottest). The final chromatographic efficiency is a net result of the two opposing mechanisms. At high flow rates in a non-thermostated system, there was a significant decrease in plate height as evident from the van Deemter plot, which was attributed to higher axial heating contributions resulting in improved efficiency (7). Though thermostating may provide higher reproducibility, it is not actively employed in most of the reported research on ultrafast chromatography, because it leads to higher extracolumn volumes.
Instrumental and Practical Considerations
It is widely accepted that chromatographic instrumentation lags behind column technology. Advances in instrumentation are as important as high-efficiency columns when it comes to ultrafast chromatography. When looking particularly at fast eluted peaks, the effect of different instrumental parameters seems to be much more profound. Achieving ultrafast separations on HPLC systems is highly difficult owing to their limitation in a variety of aspects, particularly back pressure tolerance and extracolumn volumes. Chromatographers have therefore moved on to the more developed UHPLC systems introduced commercially in 2004. Commercial UHPLC systems have come a long way since then, and current systems are equipped with highly sensitive UV detectors, lower extracolumn volumes, and better electronics, resulting in a better representation of a chromatographic system to the end user.


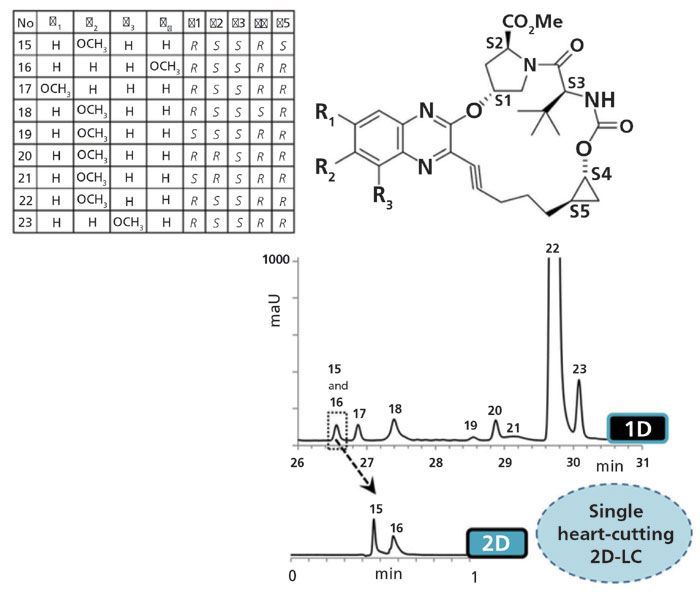
Figure 4: Single heart-cutting 2D-LC method for separation of complex mixture of closely related stereoisomers from an anti-HCV therapeutic. Conditions, first dimension (achiral): column, 150 mm × 2.1 mm, 1.6-µm Cortecs (Waters), temperature, 40 °C. Detection: UV 215 nm. Flow rate: 0.220 mL/min. Mobile phase: eluent A, 0.1% H3PO4 in H2O, eluent B, 80:20 acetonitrile-methanol (v/v). Step gradient: 20-45% B in 7 min, 7-20 min, 90% B, 20-32 min, 20% B. Sample frequency: 80 Hz. Conditions, second dimension (chiral): column, 50 mm × 4.6 mm, 1.9-µm teicoplanin (AZYP LLC), ambient temperature. Detection: UV 215 nm. Flow rate: 1.0 mL/min. Isocratic mobile phase: 5:95 0.1% H3PO4 in H2O-acetonitrile. Sampling frequency: 240 Hz. Adapted with permission from reference 26.
Ultrafast separations pose several instrumental challenges. These include the requirements of fast chromatographic detectors, high-pressure pumps with 8 mL/min flow rates, and, most importantly, an understanding of chiral column packing technology. The downside of the small particles is a decrease in column permeability, which leads to higher back pressures and the need for more sophisticated pumps to handle higher pressures and flow rates (for example, up to 800 bar and 8 mL/min).
Researchers utilized the advances in instrumentation and reported transitioning from chiral HPLC to chiral UHPLC (6), using chiral stationary phase bonded to sub-2-µm particles to produce high back pressure. Separation was performed at flow rates up to 6 mL/min to achieve ultrafast speeds and to justify the need for UHPLC systems. Researchers at Merck also managed to develop an ultrafast screening protocol on UHPLC systems for pharmaceutically important compounds using multiple columns operating under reversed-phase conditions (27). Modern-day UHPLC systems vary in the pressure that they can tolerate and flow rates up to which they can operate.
Extracolumn Band Broadening
Extracolumn band broadening stands out as a disadvantage while performing ultrafast separations. Any peak convolution process taking place outside the column, including contributions to band broadening from the injector, tubing, detector volume, and detector electronics, are deemed as extracolumn effects. Though decreasing contributions from detector volume and detector electronics are beyond a typical user's prerogative, band broadening can be significantly lowered by using narrow internal diameter (i.d.) tubing and using ultralow dispersion injectors now commercially available. A study showed a staggering 50% decrease in efficiency of the first eluted peak when tubing was changed from 75-µm i.d. tubing to regular blue PEEK tubing (254 µm i.d.) (28). A reader with the intention of performing ultrafast separations must therefore pay close attention to the choice of tubing to attain maximum efficiency from a given system.
Sampling Rates and Response Times
The most commonly used detector in LC is the diode array detector. Detector sampling rates and detector response times have become important aspects of ultrafast chromatography. Peak shapes, peak width, and baseline noise can vary considerably when changing detector settings. The sampling frequency can be varied in most HPLC and UHPLC systems. According to Shannon's theorem (21), to accurately capture an analytical signal the minimum sampling, frequency must be twice the maximum frequency components in the signal being acquired. Performing Fourier analysis on a chromatogram can extract the frequency components, and consequently uncover the minimum sampling frequency required for analysis of the analyte signal. According to the study conducted by Wahab and colleagues, by simulating a sub-second separation, current instrumentation is capable of sampling data at frequencies sufficient for commercially available high-efficiency columns, but the authors predicted that with the advent of higher efficiency columns higher sampling rates might be required to perform an accurate representation of the signal (21). The highest available sampling frequency in a UHPLC system varies across different manufacturers, some being as high as 250 Hz. This high sampling rate results in higher baseline noise and manufacturers have tried to tackle this problem by using digital filters such as "Savitzky-Golay", "Gaussian weighted moving average", "rectangular moving average", "low pass RC", and "Hamming filter" to eliminate high frequency signal components, and thereby reduce baseline noise (33). These filters are often anonymous in the instrument software. However, users can see the rise time or the response times in the detector settings of the method. Response times are defined as the time it takes for a unit step-function to go from 10% to 90% of the signal. Each digital filter can uniquely distort the peak shape if the response time is not chosen judiciously. Figure 5 shows the massive impact of sampling frequency and response times on a chromatographic signal. Only with the correct choice of both parameters can the desired chromatogram be obtained.


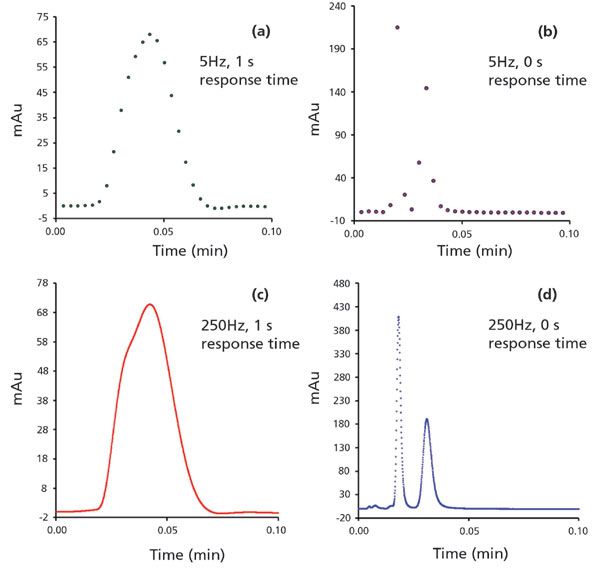
Figure 5: Ultrafast separation of oxazepam enantiomers on 2.7-µm 1 cm SPP teicoplanin column. Dependence of chromatographic signal on sampling rate and response times. The sampling rate and response times are (a) 5 Hz, 1 s. (b). 5 Hz, 0 s. (c). 250 Hz, 1 s. (d). 250 Hz, 0 s, respectively. Original work.
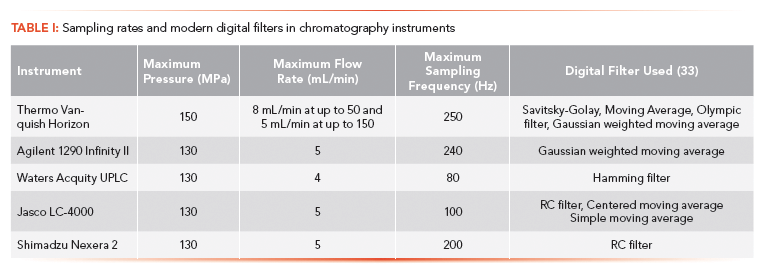
Table I gives a brief overview of a range of UHPLC instruments available from a variety of manufacturers with regards to their pressure tolerance, maximum flow rate at which they can operate, maximum sampling frequency, and the digital filter they use (data obtained from manufacturer's website).


Where do we stand in terms of speed? Following optimization of both column and instrumental parameters, chiral chromatography has reached new heights unimaginable even a few years ago. As described earlier, the time limit for an "ultrafast separation" is arbitrarily chosen to be under 1 min. This definition is sure to change in future, and as of now, sub-second chromatography is no longer a dream. Enantiomeric separations under a second have now been performed both with LC and SFC, and it has opened avenues for a variety of applications for which chromatography was earlier considered too slow (30). Pioneering work by researchers in the United States, optimizing both instrumental and practical factors, coupled with the development of packing techniques for columns as small as 0.5 cm, has led to achiral and chiral separations of a wide range of analytes under a second (21). Researchers in Italy have also reported sub-second separation using a 1-cm long pi-complex chiral selector-based column (24). Further increases in speed of chromatographic separations have recently been achieved, and as many as 10 analytes have been separated under a second (29). This unforeseen speed in the domain of chromatographic separation is sure to create a huge impact in upcoming research in this area. Figure 6 illustrates the sub-second separation of oxazepam enantiomers. Interestingly, chromatography can now be completed in a time domain that was previously limited to sensors (29).


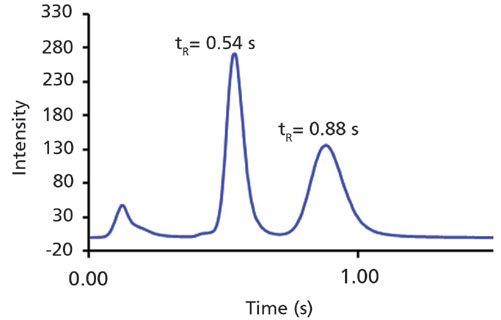
Figure 6: Sub-second separation of oxazepam enantiomers. Conditions: Column: 1 cm × 0.3 cm, 2.7-µm SPP Teicoplanin (AZYP LLC), mobile phase: 100% methanol, flow rate: 7.5 mL/min, UV-vis detection at 254 nm. Original work.
Conclusions
The progress of enantiomeric separations, particularly with regards to speed, has been astonishing in the last few years. The dream of ultrafast chiral separation has been realized by groundbreaking work by researchers both in industry and academia. Commercial availability of sub-2-µm FPP and 2.7-µm SPP-based chiral stationary phases will further accelerate research in the domain of ultrafast chiral separations (21,29,31,32). Since most chiral stationary phases used in LC can also be used in SFC, both the techniques will benefit from these advances. Owing to the tremendous progress in this area, the need for better instrumentation seems to be imminent. Finally, as these developments result in even faster separations, the impact will be felt in a multitude of diverse fields, such as monitoring short lived intermediates and on-line reaction profiles, and even more so if the entire system can be miniaturized.
References
1) C.G. Horvath, B. Preiss, and S.R. Lipsky, Anal. Chem. 39, 1422–1428 (1967).
(2) D. Randau and W. Schnell, JJ. Chromatogr. A 57, 373–381 (1971).
(3) Z. Liu and J.B. Phillips, J. Microcolumn Sep. 1, 249–256 (1989).
(4) F. Gasparrini, D. Misiti, and C. Villani, TrAC, Trends Anal. Chem. 12, 137–144 (1993).
5) T.J. Ward and K.D. Ward, Encyclopedia of Analytical Chemistry (John Wiley & Sons, Hoboken, New Jersey, 2000).
(6) D. Kotoni, A. Ciogli, C. Molinaro, I. D'Acquarica, J. Kocergin, T. Szczerba, H. Ritchie, C. Villani, and F. Gasparrini, Anal. Chem.84, 6805–6813 (2012).
(7) D.C. Patel, Z.S. Breitbach, M.F. Wahab, C.L. Barhate, and D.W. Armstrong, Anal. Chem. 87, 9137–9148 (2015).
(8) E.L. Regalado and C.J. Welch, J. Sep. Sci.38, 2826–2832 (2015).
(9) J.J. Van Deemter, F. Zuiderweg, and A.V. Klinkenberg, Chem. Eng. Sci. 5, 271–289 (1956).
(10) R. Hayes, A. Ahmed, T. Edge, and H. Zhang, J. Chromatogr. A 1357, 36–52 (2014).
(11) P. Carman, Chem. Eng. Res. Des. 75, S32–S48 (1997).
(12) G. Cancelliere, A. Ciogli, I. D'Acquarica, F. Gasparrini, J. Kocergin, D. Misiti, M. Pierini, H. Ritchie, P. Simone, and C. Villani, J. Chromatogr. A1217, 990–999 (2010).
(13) I. Halász, R. Endele, and J. Asshauer, J. Chromatogr. A 112, 37–60 (1975).
(14) D. Cabooter, A. Fanigliulo, G. Bellazzi, B. Allieri, A. Rottigni, and G. Desmet, J. Chromatogr. A 1217, 7074–7081 (2010).
(15) M. Catani, O.H. Ismail, A. Cavazzini, A. Ciogli, C. Villani, L. Pasti, C. Bergantin, D. Cabooter, G. Desmet, and F. Gasparrini, J. Chromatogr. A1454, 78–85 (2016).
(16) C.L. Barhate, M.F. Wahab, Z.S. Breitbach, D.S. Bell, and D.W. Armstrong, Anal. Chim.Acta 898, 128–137 (2015).
(17) O.H. Ismail, A. Ciogli, C. Villani, M. De Martino, M. Pierini, A. Cavazzini, D.S. Bell, and F. Gasparrini, J. Chromatogr. A 1427, 55–68 (2016).
(18) M.F. Wahab, D.C. Patel, R.M. Wimalasinghe, and D.W. Armstrong, Anal. Chem. 89, 8177–8191 (2017).
(19) K. Lomsadze, G. Jibuti, T. Farkas, and B. Chankvetadze, J. Chromatogr. A 1234, 50–55 (2012).
(20) D.A. Spudeit, M.D. Dolzan, Z.S. Breitbach, W.E. Barber, G.A. Micke, and D.W. Armstrong, J. Chromatogr. A1363, 89–95 (2014).
(21) M.F. Wahab, R.M. Wimalasinghe, Y. Wang, C.L. Barhate, D.C. Patel, and D.W. Armstrong, Anal. Chem. 88, 8821–8826 (2016).
(22) D.C. Patel, Z.S. Breitbach, J. Yu, K.A. Nguyen, and D.W. Armstrong, Analytica Chimca Acta 963, 164–174 (2017).
(23) Y. Min, Z. Sui, Z. Liang, L. Zhang, and Y. Zhang, J. Pharm. Biomed. Anal. 114, 247–253 (2015).
(24) O.H. Ismail, L. Pasti, A. Ciogli, C. Villani, J. Kocergin, S. Anderson, F. Gasparrini, A. Cavazzini, and M. Catani, J. Chromatogr. A1466, 96–104 (2016).
(25) O.H. Ismail, M. Antonelli, A. Ciogli, C. Villani, A. Cavazzini, M. Catani, S. Felletti, D.S. Bell, and F. Gasparrini, J. Chromatogr. A 1520, 91–102 (2017).
(26) C.L. Barhate, E.L. Regalado, N.D. Contrella, J. Lee, J. Jo, A.A. Makarov, D.W. Armstrong, and C.J. Welch, Anal. Chem. 89, 3545–3553 (2017).
(27) C.L. Barhate, L.A. Joyce, A.A. Makarov, K. Zawatzky, F. Bernardoni, W.A. Schafer, D.W. Armstrong, C.J. Welch, and E.L. Regalado, Chemical Communications 53, 509–512 (2017).
(28) D.C. Patel, M.F. Wahab, D.W. Armstrong, and Z.S. Breitbach, J. Chromatogr. A 1467, 2–18 (2016).
(29) D.C. Patel, M.F. Wahab, T.C. O'Haver, and D.W. Armstrong, Anal. Chem. 90, 3349–3356 (2018).
(30) A. Ciogli, O.H. Ismail, G. Mazzoccanti, C. Villani, and F. Gasparrini, J. Sep. Sci. 41, 1307–1318 (2018).
(31) G. Hellinghausen, D. Roy, Y. Wang, J.T. Lee, D.A. Lopez, C.A. Weatherly, and D.W. Armstrong, Talanta 181, 132–141 (2018).
(32) C.L. Barhate, D.A. Lopez, A.A. Makarov, X. Bu, W.J. Morris, A. Lekhal, R. Hartman, D.W. Armstrong, and E.L. Regalado, J. Chromatogr. A 1539, 87–92 (2018).
(33) M.F. Wahab, P.K. Dasgupta, A.F. Kadjo, and D.W. Armstrong, Anal. Chim. Acta 907, 31–44 (2016).
Daipayan Roy is a PhD candidate at the University of Texas at Arlington. His research interests include high throughput chiral and achiral separations using HPLC, HILIC, SFC, and MS.
Choyce Weatherly is a research engineering scientist at the University of Texas at Arlington. He completed his Ph.D. at the University of Texas at Arlington. His research includes method development, data analysis, and chiral–achiral separation techniques using HPLC, HPLC–MS, GC, GC–MS, and SFC.
M. Farooq Wahab is currently a research engineering scientist at the University of Texas at Arlington. He undertook a postdoctoral fellowship with Professor Armstrong after completing his Ph.D. at the University of Alberta, Canada. His work is based on understanding the fundamentals of separation science, signal processing for chromatography, ion chromatography, HILIC, SFC, and ultrafast LC.
Daniel W. Armstrong is the Welch Distinguished Professor of Chemistry at the University of Texas at Arlington. Professor Armstrong has received more than 30 national and international research and teaching awards. His current interest involves chiral recognition, macrocycle chemistry, synthesis and use of ionic liquids, separation science, and mass spectrometry. He has more than 600 publications, one book, and 30 patents. Direct correspondence to sec4dwa@uta.edu.










In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




