Biomimetic Chromatography to Accelerate Drug Discovery: Part II
LCGC North America
Biomimetic HPLC retention data can be used to measure how a compound will bind to proteins and phospholipids in vivo.
The high performance liquid chromatography (HPLC) measurements of the biomimetic properties of drug discovery compounds has been described and reviewed previously (1). The property measurements are based on calibrated generic gradient retention times obtained on C18, human serum albumin (HSA), alpha-1-acid-glycoprotein (AGP), and immobilized artificial membrane (IAM) stationary phases.
As the gradient retention times are dependent on various HPLC conditions, such as flow rate, dwell volume of the instrument, and column dimensions, it is very important to calibrate the gradient retention times with a set of compounds for which the percentage of binding data is available. In this way, the gradient retention times obtained on one HPLC system can be converted to another, and the data can be collated in a database suitable for interlaboratory comparison. It is advisable to measure the retention times of the calibration set of compounds on a daily basis. The slope and intercept of the calibration lines are determined using the predefined binding constants. By using the actual slope and intercept values on a specific day and on a particular instrument and column combination, the gradient retention times can be converted to binding constants or to chromatographic hydrophobicity indices (CHIs) that are suitable for interlaboratory comparison, and can be used in the model equations described in this article.
Using the Abraham solvation equation approach (2), differences between the lipophilicity of compounds measured by octanol–water, reversed-phase retention, and biomimetic binding data are identified. The major differences between the systems are their sensitivity toward the H-bond acidity descriptor. Compounds with H-bond donor groups partition to octanol easily, similarly to the IAM and HSA partition, because they can form hydrogen bonding in the nonaqueous phase. They are not sensitive toward H-bond donor groups. The similarity between octanol–water partition and IAM–HSA binding is valid only for neutral molecules. While the octanol–water partition coefficients and the CHI drop when molecules are ionized, regardless of the type of charge, the IAM binding will be increased by the presence of a positive charge and the albumin binding will be increased by the presence of a negative charge. The conclusion was that the traditionally used octanol–water lipophilicity is different from the biomimetic HPLC partition coefficients. Therefore, it is essential to investigate how the biomimetic properties relate to in vivo distribution properties of compounds.
Modeling In Vivo Steady State Volume of Distribution (Vdss)
The volume of distribution in the steady state (Vdss) can be measured only in vivo. It is defined as the dose over the steady state plasma concentration (cplasma) as shown in equation 1.



The volume of distribution is usually derived from the plasma concentration versus time profile of the compound. It can be derived mathematically and is proportional to the tissue–plasma partition coefficient, K, that is, the concentration of the compound in tissues divided by the concentration of the compound in plasma (equation 2):


where Vp and Vt are the volume of the plasma and the tissue, respectively. Tissues contain phospholipids, while the plasma does not. It can be assumed that the difference between the phospholipid (IAM) and albumin (HSA) binding drives the compounds into the tissue compartment. The human clinical steady-state volume of distribution data of over 150 marketed drug molecules has been modeled by the measured IAM and HSA binding of the compounds (3). Equation 3 shows the model and Figure 1 shows the plot that was obtained by plotting the human clinical volume of distribution in the function of the predicted value using equation 3.

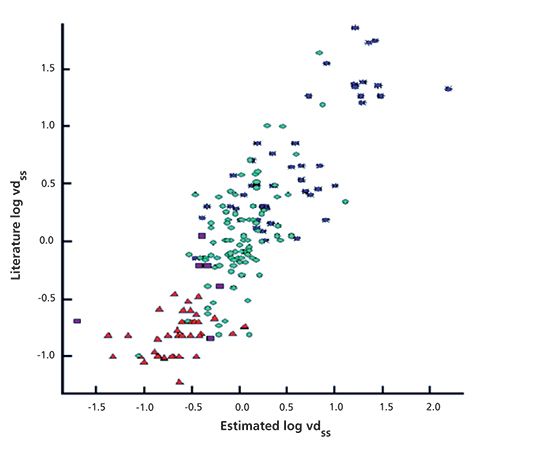
Figure 1: The measured and predicted human clinical steady-state volume of distribution (log Vdss) of over 150 marketed drugs. Adapted with permission from reference 3.


Note that the log K (IAM) and log K (HSA) is scaled to the log P octanol–water scale, so that the values can be compared directly with each other. The log K (IAM) can be calculated from the log k (IAM) derived from the CHI IAM values using equation 4, while the log K (HSA) can be derived from the log k (HSA) values obtained from the correlation to the log retention times using equation 5.


Since positively charged compounds bind stronger to IAM while negatively charged compounds bind stronger to albumin, it can be concluded that it is the charge that drives the compound out from the plasma compartment or keeps it in.
A good example of the superiority of the biomimetic binding measurements over the octanol–water lipophilicity is shown in Figure 2. Nifedipine and amlodipine have very similar structures. Amlodipine has an aliphatic primary amino group that makes the compound basic and positively charged at physiological pH. Nifedipine is a neutral molecule without charge. The octanol–water log P is higher for nifedipine than amlodipine, while the log D of amlodipine at pH 7.4 drops significantly, because of the presence of the positively charged primary amino group. However, amlodipine has a much larger volume of distribution, and a much longer half-life than nifedipine, which is a result of strong tissue binding. While the octanol–water lipophilicity values do not reflect these differences, the biomimetic albumin and IAM binding data suggest a high volume of distribution for amlodipine.

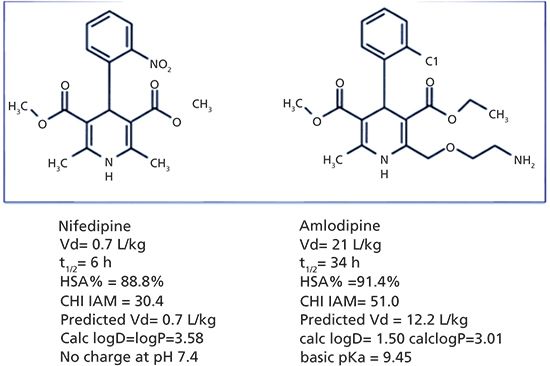
Figure 2: The in vivo human volume of distribution, half-life data, and the measured biomimetic HSA and IAM binding data for nifedipine and amlodipine. The estimated volume of distribution of amlodipine using equation 3 is much higher than was estimated for nifedipine.
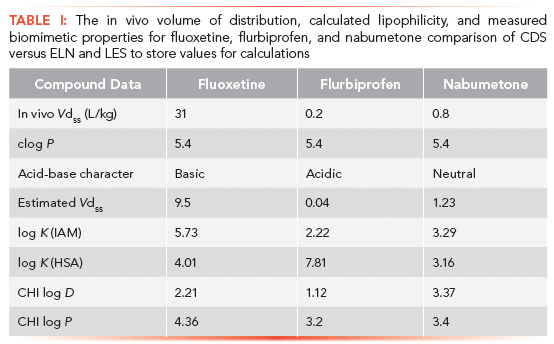
The log P values of fluoxetine, flurbiprofen, and nabumetone are almost the same, suggesting the same lipophilicity for the three drug molecules. However, their measured and estimated volumes of distribution are very different. The biomimetic HSA and IAM binding data accurately predicted that. The major differences between these compounds are in their charge state, fluoxetine being basic, flurbiprofen acidic, and nabumetone neutral. Figure 3 shows the structures, and Table I shows the measured volume of distribution and protein binding together with the measured CHI lipophilicity, IAM, and HSA binding data for fluoxetine, flurbiprofen, and nabumetone.

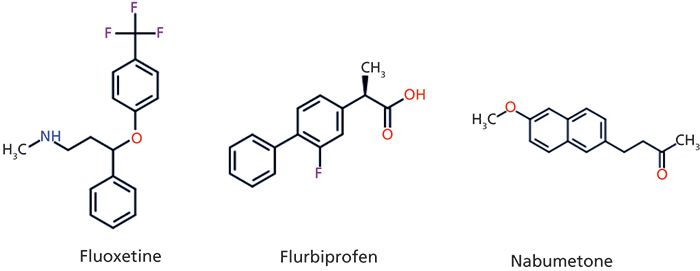
Figure 3: The structures of fluoxetine, flurbiprofen, and nabumetone as examples for basic, acidic, and neutral drugs. The measured and estimated volumes of distribution data together with the biomimetic and lipophilicity data are shown in Table I.
It can also be seen in Table I that the measured phospholipid binding would not predict the low volume of distribution of nabumetone on its own, and the phospholipid binding should always be compared to the albumin binding. A compound binds more to IAM relative to its albumin binding when the in vivo volume of distribution is high. The simple lipophilicity data, either octanol–water or the C18 reversed-phase lipophilicity, are not able to indicate the extent of the volume of distribution of a compound.

The model described by equation 3 is based on the human clinical volume of distribution data of marketed drugs. A similar model has been set up for estimating the volume of distribution in a rat. The data for 247 drug discovery compounds from GSK (3) were used to build a model for the rat volume of distribution as shown by equation 6:


where n is the number of compounds, r2 is the multiple regression coefficient, s is the standard error of the estimate, and F is the Fisher-test value from the significance test.
When both the IAM and the HSA binding data are converted to the log P scale, the regression coefficients of equations 3 and 6 are comparable and meaningful. It shows that the IAM coefficient in the rat model is much smaller than in the human model, indicating that the rat has smaller quantities of phospholipids in the body relative to the plasma protein (albumin). There is, therefore, a possibility to use the biomimetic binding data as input into physiologically based pharmacokinetic modeling (4,5).
In conclusion, therefore, the biomimetic IAM and HSA binding can be used to estimate the human clinical volume of distribution.
Estimating In Vivo Unbound Volume of Distribution (Vdu)
It has been recognized that the unbound drug molecules can interact with the receptors, so it is necessary to optimize the unbound concentration of the drug in the body. The volume of distribution unbound (Vdu) is defined as the proportion of the dose and the free plasma concentration (cfree) as described by equation 7.


This can be calculated from the steady state volume of distribution and the unbound fraction (fu) of the drug in plasma, as cfree = fu × cplasma, and Vdu equals Vd/fu.
When modeling the unbound volume of distribution, it was found that the sum of the two types of binding that influence the unbound volume of distribution could be described by equation 8.

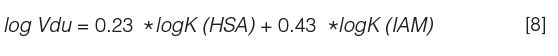
Figure 4 illustrates the difference between the volume of distribution and the unbound volume of distribution. While the optimum volume of distribution of a drug molecule is not known, it is possible to aim for a low unbound volume of distribution because a low unbound volume of disruption means high free concentration relative to dose (see equation 7). For some drugs where the target is in the tissues, it is advantageous for the compound to partition extensively into the tissues to have a higher concentration at the site of action. For example, for drugs entering the central nervous system (CNS), the volume of distribution is often higher than for non-CNS drugs (Figure 5).

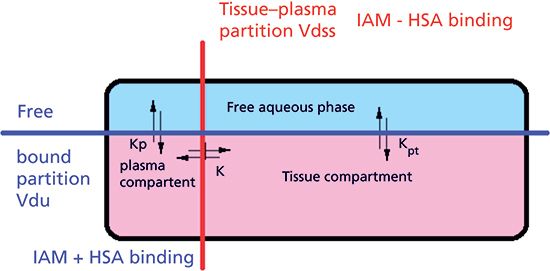
Figure 4: The schematic illustration of the equilibrium processes of drug molecules in the plasma and the tissue compartment.
The unbound volume of distribution should however always be low regardless of where the drug target is in the body. This becomes quite clear when we think of the recently introduced drug efficiency concept (DRUGeff) of Braggio and coauthors (6).

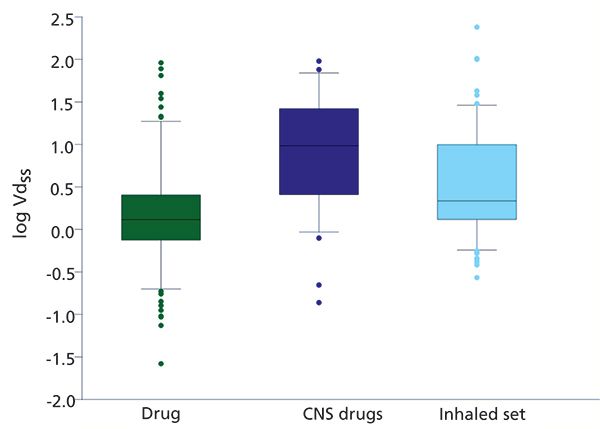
Figure 5: The average volume of distribution of drugs, CNS drugs, or drugs showing CNS side effects and inhaled drugs.
Estimating In Vivo Drug Efficiency by Biomimetic HPLC Data
According to the drug efficiency concept, a drug is more efficient when it achieves high free concentration at the site of action with low dosage. It is defined by equation 9.


It is very difficult to measure the free biophase concentration, especially when there is an intracellular target. It is common to estimate the free concentration in the tissues from the free concentration of the drug in plasma. This assumption is valid when the free drug hypothesis is true. The free plasma concentration relative to dose provides the maximum drug efficiency (DEmax). It can be achieved assuming the compounds have 100% bioavailability, no active transport to distort the thermodynamic equilibrium in different compartments, and there is no permeability barrier preventing the compound from going through the cell membrane. This is the best case scenario in terms of drug efficiency. In this case the maximum drug efficiency can be considered as the reciprocal value of the Vdu. Figure 6 shows the agreement between the in vivo DEmax values and the estimated values using the model for Vdu described by equation 8 and taking the reciprocal value of the estimated Vdu values (7).

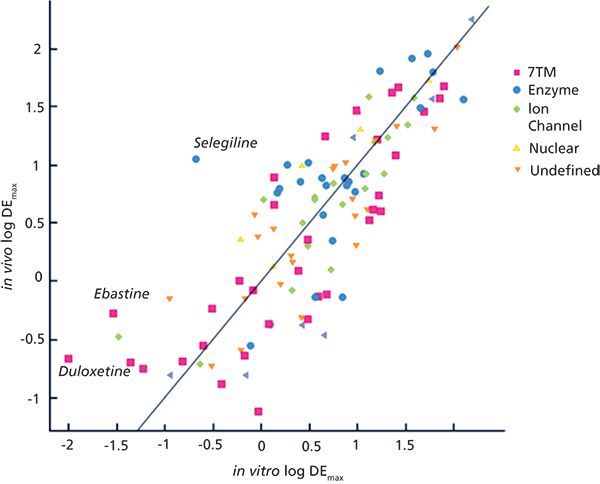
Figure 6: The estimated DEmax using the biomimetic HSA and IAM binding and the in vivo DEmax for marketed drugs acting on various targets. Adapted with permission from reference 7.
For many therapeutic areas, the in vivo drug efficiency max could be modelled by the in vitro drug efficiency max within a 1 log unit error, as shown in Figure 6. The line in Figure 6 is not a regression line, but a line of unity. Some drugs are outliers, such as selegiline, ebastine, and duloxetin. Selegiline is an irreversible inhibitor of type B mono-amino-oxidase, so in this case the free plasma concentration is not relevant. As it irreversibly binds to the enzyme it has higher local concentration than predicted from the in vitro binding measurements. Duloxetin is a serotonin re-uptake inhibitor and the clinical effect can be observed only after several weeks of treatment, probably after saturation of several nonspecific binding sites. Ebastine shows much higher in vivo free concentration than predicted from its in vitro binding properties, which could be a result of some active transport properties.
The most significant application of DEmax in early drug discovery is when it is combined with in vitro potency (expressed as the negative logarithm of the concentration of the compound causing 50% inhibition in the in vitro potency assay, called pIC50) to obtain the drug efficiency index (DEI) described by equation 10.


It has been shown that DEI is proportional to the receptor occupancy per unit of dose (8). It is also useful for ranking early drug discovery compounds based on the expected in vivo potency. When the DEmax is less than 1% the log DEmax becomes a negative number, which is taken away from the potency. Compounds with DEmax values greater than 1 show higher potency in vivo. Known drug molecules have a DEmax above 1%. When the pIC50 and the DEI values are plotted for known drugs, they are around a line of unity, which means a DEmax of around 1%. However, drug discovery project compounds may show similar in vitro potency, but a range of DEmax values. The most developable compounds are those that have both high potency and high drug efficiency (Figure 7). The green points show the marketed drugs, and the yellow points are the project compounds showing all pIC50 between 6 and 8. Compounds marked as red stars are the selected candidate molecules based on their advantageous ADME properties and in vivo efficacy.

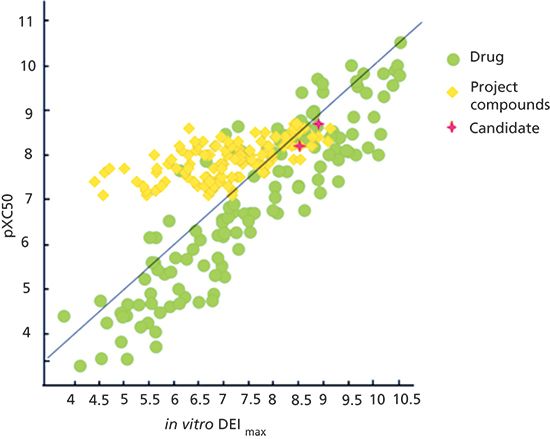
Figure 7: The plot of pIC50 and DEI values for marketed drugs (green), project compounds (yellow), and selected candidate molecules (red). Adapted with permission from reference 7.
The question is whether the same behavior and ranking could be predicted simply by using the lipophilicity of the compounds that are usually expressed as octanol–water partition coefficients. As we have seen in Table V of the previous article (1), both the phospholipid binding and albumin binding are similar to the octanol–water partition when the solvation equation is used to relate them to the molecular properties. The drug efficiency is inversely related to the sum of HSA and IAM binding, therefore it can be expected that this sum is proportional to the octanol–water log P and not the log D for ionized compounds. When the compound is negatively charged, it binds more strongly to albumin, while positively charged compounds bind more strongly to the IAM stationary phase. Figure 8 was obtained when plotting the DEmax values of marketed drugs as a function of their clog P values. A reasonably good inverse correlation can be seen between the log DEmax and clog P values of the marketed drugs. Positively charged and negatively charged compounds are well integrated and not separated in the plot.

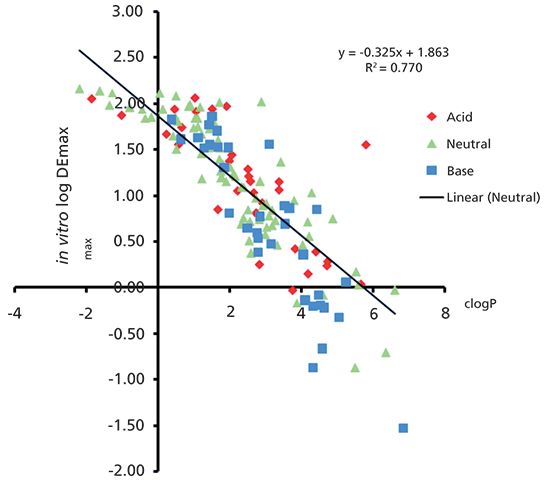
Figure 8: The plot of DEmax (sum of the HSA and IAM binding) of marketed drugs versus their clog P values.
The differences are relatively large caused by the 3D contribution from the measured binding properties, while the clog P values are in silico calculations using 2D descriptors only. The albumin binding is selective and can differentiate between enantiomers, while the octanol–water system cannot. It is important to emphasize that during the biomimetic binding measurements, stationary phases that are the components of the body (albumin, glycoprotein, phosphatidylcholine) are used. Dynamic equilibrium is measured between the aqueous mobile phase at physiological pHs on the large stationary phase surface. The biological distribution processes also take place on large surfaces of the cell membrane, or proteins in a dynamic way, so that the biomimetic HPLC process should be more similar to the biological distribution than the liquid–liquid partition process in the octanol–water partition. It may be difficult to obtain accurate affinity partition coefficient values by HPLC. However, the reproducibility can be improved by using a standard set of compounds with known binding values to calibrate the actual HPLC system. The ranking order of the binding is very reproducible, and because of the high resolution of the HPLC system it can differentiate between closely related compounds.
Modeling Brain Tissue and Lung Tissue Binding with Biomimetic HPLC Data
As tissues are composed of proteins and lipids, it is possible to estimate tissue binding of compounds from their biomimetic HPLC binding data. The brain tissue binding model was derived using 135 drug discovery research compounds using equilibrium dialysis data (8). Equation 11 describes the model. Note, in this case the IAM and HSA binding data are not scaled to the octanol–water partition coefficients, so the regression coefficients do not reflect the tissue composition accurately. The brain tissue binding (BTB) was expressed as the log partition coefficients between the free and tissue bound concentration derived from the equilibrium dialysis and expressed as log k (BTB).


where n is the number of compounds, r2 is the multiple regression coefficient, s is the standard error of the estimate, and F is the Fisher significance test value. The plot between the measured and estimated log k (BTB) values are shown in Figure 9. Similarly, good correlation was found between the lung tissue binding and the biomimetic HSA and AGP binding data. Interestingly, the mucus binding showed excellent correlation to the AGP binding because mucus contains 90% glycoproteins. The AGP binding may become important in modeling lysosomal binding (trap) because lysosomes contain a large percentage of negatively charged glycoproteins, causing lower pH around the lysosomes, which attracts positively charged compounds just like AGP.

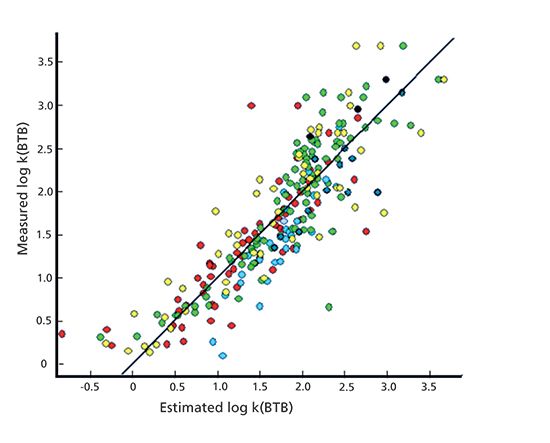
Figure 9 The plot of the measured brain tissue binding by equilibrium dialysis log k (BTB) versus the estimated log k (BTB) values using the biomimetic HSA and IAM binding data.
Conclusions
This article demonstrates the applicability of biomimetic HPLC retention data for measuring a compound's binding to proteins and phospholipids. The fast generic HPLC retention times obtained using protein and immobilized artificial membrane stationary phases can be used to derive a compound's affinity to proteins and phospholipids. In order to obtain binding data suitable for interlaboratory comparisons and building of in vivo models, a calibration set of compounds would have to be used and their gradient retention data measured. The calibration line obtained from the gradient retention times and the fixed binding values can be used to calculate the binding of drug discovery compounds. The measurements can be automated and take only 6 min on a chromatographic system. These measurements have been carried out on over half a million research compounds and have been used to select candidate molecules. The models were developed and validated using the data comparison on marketed drugs and have been published (8). The calibrated gradient retention times are very reproducible and because of the high resolution of the chromatographic system it is easy to build structure–property relationships.
References
(1) K. Valko, LCGC North Am. 36(6), 397–405 (2018).
(2) M.H. Abraham, H.S. Chadha, G.S. Whiting, and R.C. Mitchell, J. Pharm.Sci. 83, 1085–1100 (1994).
(3) F. Hollósy, K. Valkó, A. Hersey, S. Nunhuck, G. Kéri, and C. Bevan, J. Med. Chem. 49, 6958–6971 (2006).
(4) H.M. Jones, N. Parrott, K. Jorga, and T. Lavé, Clin. Pharmacokinet. 45, 511–542 (2006).
(5) P. Poulin and F.-P. Theil, J. Pharm.Sci.91, 1358–1370 (2002).
(6) S. Braggio, D. Montanari, T. Rossi, and E. Ratti, Expert Opin. on Drug Discovery 5, 609–618 (2010).
(7) K. Valko, E. Chiarparin, S. Nunhuck, and D. Montanari, J. Pharm.Sci. 101, 4155–4169 (2012).
(8) K. Valko, Physicochemical and Biomimetic Properties in Drug Discovery - Chromatographic Techniques for Lead Optimization, (John Wiley & Sons, Hoboken, New Jersey, 2014).
Klara Valko is founder and director of Bio-Mimetic Chromatography Ltd. She is an Honorary Professor at the UCL School of Pharmacy since 2004 and leads the Physchem/ADME module for the Drug Discovery MSc course. Direct correspondence to: Klara_Valko@bio-mimetic-chromatography.com










In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




