Forensic Drug Screening by LC–MS Using Accurate Mass Measurement
LCGC Europe
Because of progress in liquid chromatography/time-of-flight mass spectrometry (LC/TOF-MS) instrumentation, data processing and reporting, the measurement of compounds' accurate masses is becoming routine practice in screening analysis based on target databases. As such databases of monoisotopic masses can be easily updated with recent data from the literature; rapid characterization of new compounds and metabolites is possible without the need for primary reference standards. This approach has already been established in comprehensive toxicological urine screening and in analysis of drugs-of-abuse in seized street drug samples. Currently, a mass accuracy within 5 ppm can be routinely achieved, and confirmation via a numerical isotopic pattern match (SigmaFit) is provided by a new generation LC/TOF-MS instrument.
Because of progress in liquid chromatography/time-of-flight mass spectrometry (LC/TOF-MS) instrumentation, data processing and reporting, the measurement of compounds' accurate masses is becoming routine practice in screening analysis based on target databases. As such databases of monoisotopic masses can be easily updated with recent data from the literature; rapid characterization of new compounds and metabolites is possible without the need for primary reference standards. This approach has already been established in comprehensive toxicological urine screening and in analysis of drugs-of-abuse in seized street drug samples. Currently, a mass accuracy within 5 ppm can be routinely achieved, and confirmation via a numerical isotopic pattern match (SigmaFit) is provided by a new generation LC/TOF-MS instrument.
Introduction
Screening for drugs and poisons is an essential part of forensic sciences, clinical toxicology and doping control.1,2 Depending on the purview of the laboratory, the analytical procedure may be targeted solely to the major drugs-of-abuse or may cover all scheduled drugs. In forensic toxicology, it is often necessary to reliably screen for hundreds or thousands of toxicologically relevant substances, a task which is particularly demanding. Although a key factor in analytical toxicology, substance identification has received inadequate attention. To improve the situation, uniform analytical strategies, computerized databases and banks of reference standards have been proposed.3
While blood concentration best reflects the acute action of a substance, screening analysis is often performed in urine because it offers a wider time window of detection than blood. For hydrophilic compounds, the concentration at any given moment is usually higher in urine, making detection more reliable. Lipophilic compounds, by contrast, exist in urine mainly as metabolites, which are more hydrophilic than the parent compound. If metabolite analysis is available, the metabolic patterns can be used to aid identification. An exact urine drug concentration usually has a very limited interpretative value, but a cut-off concentration differentiating between positive and negative findings should be specified, particularly for drugs-of-abuse.
No single analytical technique has to date shown an overwhelming predominance in comprehensive screening analysis; resolving a case usually requires the proper use of a multitude of techniques. Naturally, the technique itself does not guarantee a successful outcome but rather the efficiency of the method used. A standardized procedure for gas chromatography/mass spectrometry (GC–MS) in the electron impact full-scan mode, which uses large spectral libraries,4 has become established in urine drug screening within the last two decades. While this technique is sometimes referred to as the gold standard, it has limitations in terms of sensitivity, specificity and scope. These have been anticipated to be overcome by applying liquid chromatography/mass spectrometry (LC–MS). Very recently, the focus of LC–MS in screening analysis has been in accurate mass measurement.
Drug Screening by LC–MS
The problems of existing screening techniques, such as the compound volatility requirements in gas chromatography and the moderately non-selective diode array detection in liquid chromatography, have made LC–MS particularly appealing to forensic drug analysts. Most methods developed have, however, targeted a limited number of analytes, typically one drug and its main metabolites. Comprehensive screening methods started to emerge in scientific literature only ten years ago.5
Several types of LC–MS techniques based on single or triple quadrupole, ion-trap and time-of-flight (TOF-MS) mass analysers have been applied to drug screening.6 Single quadrupole LC–MS with in-source collision-induced dissociation (CID) has led to the development of comprehensive library-based screening methods.7–9 Typically, positive and negative spectra are created simultaneously at high and low orifice voltage, showing extensive and weak fragmentation, respectively, and the spectra are summed at both polarities to produce informative spectral libraries. Because single MS methods suffer from a lack of specificity in distinguishing co-eluting substances, emphasis ought to be directed towards chromatographic resolution. Furthermore, there may be difficulty in harmonizing the CID conditions to yield reproducible spectra.10
More reliable identification can be obtained by tandem MS (LC–MS–MS) with triple quadrupole,11 quadrupole/ion-trap12 or quadrupole/TOF technology13 because the product ion spectra generated by these techniques are less dependent on sample composition and experimental settings. Recently, LC–MS–MS libraries created with different manufacturers' instruments under standardized conditions were shown to be sufficiently compatible for interlaboratory use.14 Another mode of operation within LC–MS–MS that has been applied to screening analysis is multiple reaction monitoring (MRM).15 It is a more sensitive mode of operation, but there is a technical limit for the number of target compounds that can be monitored by MRM-type experiments, hindering the utility of this approach.
The disadvantages of LC–MS screening methods are related to the commonly used ionization techniques of atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI). Volatile buffers are required for chromatography, which limits the optimization of separation by the mobile phase. In addition, neither technique applies to all types of compounds. Especially where ESI is involved, the risk for occasional ion suppression16 and consequently false negative findings always exists in comprehensive screening analysis, where matrix effects can not be controlled for the numerous library compounds.
Advantages of Accurate Mass Measurement
Accurate mass has been used in GC–MS target screening for environmental pollutants by magnetic sector instruments for 30 years already, and this technique remains in use today.17 More recently, the field of combinatorial chemistry related to drug discovery created a need for rapid characterization of the complex mixtures generated by synthesis. LC coupled to either modern Fourier transform ion cyclotron MS (FT-MS)18 or orthogonal acceleration TOF-MS19 made routine accurate mass measurement of even thermally unstable and higher molecular mass compounds feasible. Currently, LC/TOF-MS is the most cost-effective technique for performing accurate mass analysis of small molecules on a routine basis.20 In addition to high mass accuracy, the benefits of TOF-MS include medium resolution, wide mass range and fast mass spectral acquisition speed with high full-scan sensitivity, all attributes being superior to those obtained with a scanned quadrupole. Consequently, LC/TOF-MS methods have found extensive use in analytical research in, for example, the structure elucidation of metabolites,21 pesticides22 and steroids.23 However, comprehensive screening analysis, that is substance monitoring against a large target database, has not been routine until now, mainly because of limitations in data acquisition and processing.
Urine Drug Screening by LC/TOF-MS
A few years ago our group at the Department of Forensic Medicine, University of Helsinki, introduced a novel urine drug screening method using LC/TOF-MS.24,25 The approach was seminal in using the principle of accurate mass measurement for high-throughput comprehensive drug screening in a biological matrix. Sample preparation consisted of solid-phase extraction of hydrolysed urine samples. Identification of drugs, metabolites and pesticides was based on their accurate mass, retention times if a reference substance was available and drug metabolite patterns in urine. The database consisted of more than 600 compounds and new compounds could be simply added when elemental formulae of the substances were known. From the LC/TOF-MS acquisition data, the automated target database search reported hits within a ±30 ppm mass tolerance, a retention time window of ±0.2 min and a minimum area count of 500.



Table 1: Results for the case in Figure 1, produced by a visual basic macro programme. The parent drug and metabolites are reported together according to their compound codes. Some metabolites were identified by accurate mass only, based on the existing parent drug, and thus have no retention time error.
The mean mass accuracy for correct findings was 7 ppm, however, the mass window was set to 30 ppm to ensure that no interesting candidates were lost. For evaluation of the method, 50 authentic urine samples were analysed and positive findings confirmed by GC–MS. Only a few false positives were because of LC/TOF-MS. By contrast, several apparently correct findings of metabolites were missed by GC–MS. Advantages of the LC/TOF-MS method as compared with conventional screening techniques are good sensitivity, ability to screen a large number of drugs simultaneously and extreme flexibility in updating the target database library. At present, the method is an essential part of our daily routine analysis of drugs and poisons in urine. It has revealed a number of drugs-of-abuse, such as amphetamine, buprenorphine, fentanyl, LSD, methylenedioxymethamphetamine and morphine, that traditionally require a dedicated method, as well as ubiquitous prescription drugs and metabolites.
Figure 1 shows a typical complex total ion chromatogram of a urine sample run by the present method. It is noteworthy that the target compounds do not necessarily appear as distinct peaks, but they still give clear extracted ion chromatograms. Table 1 shows a report for the above situation, after refining the extensive amount of data to an easily interpretable form. The compounds without a retention time in the report are those for which reference substances were not available, being mainly metabolites and designer drugs. Thiethylperazine is one of the few recurrent false positive findings of which the analyst must be aware. Dibenzepin is added to urine samples as an extraction and chromatographic standard. The 3 to 5 digit numerical code for each compound expresses the compound group, the number of compounds within the group and the ordinal number of the compound in the group. If the metabolic profile for a compound is unknown or not used, the compound code is denoted with n/a.


Figure 1: LC/TOF-MS total ion chromatogram of a solid-phase extracted urine sample. The sample matrix can be complex and the target compounds do not necessarily appear as distinguishable peaks.
Towards More Accurate Mass Measurement
As modern LC/FT-MS instruments are capable of producing the highest mass accuracy and resolution, the technique is very attractive for the identification of drugs in biological samples. We compared our established LC/TOF-MS method with LC/FT-MS by analysing three authentic urine samples collected at autopsy.26 The LC/FT-MS was capable of producing mass accuracy within 3 ppm, in contrast to the LC/TOF-MS, which required a 20 ppm mass window. The mass accuracy of the LC/FT-MS was less affected by internal mass scale calibration than the LC/TOF-MS, for which errors using external mass calibration varied from 35 to 107 ppm. The 3 ppm mass accuracy proved to be sufficient to elucidate the elemental formula against a large toxicology target library consisting of 7640 theoretical masses.27 Disadvantages of the LC/FT-MS methodology are high cost of purchase and demanding maintenance and operation.
We evaluated one of the new generation LC/TOF-MS instruments (Bruker Daltonik GmbH) for drug screening. In a preliminary study, the mean and median mass error for drug components in a typical urine sample was 3 ppm and 1 ppm, respectively, suggesting a significant improvement over our established method using an older generation instrument. Figure 2 relates the mass windows applicable for urine drug analysis by various MS techniques to the corresponding average number of potential drug candidates in the toxicology library above. Regardless of whether the window size is 5 ppm or 3 ppm, the average number of potential library compounds is two in a library of this size.26


Figure 2: Comparison of three types of instruments for identification by accurate mass: an older generation TOF-MS, a new generation TOF-MS and an FT-MS. Based on authentic urine samples containing drugs and their metabolites, a mass window estimated for each instrument is related to the corresponding number of possible drug candidates in a toxicology library of 7640 compounds.
Drug Analysis without Primary Reference Standards
Limited availability of reference substances, especially for new drugs and metabolites, is a drawback with conventional analytical techniques. In a recently published paper from our laboratory,28 designer drugs were identified in seized samples using the established LC/TOF-MS method without reference substances. In all, 21 seized samples were analysed blind, and the results were compared with accredited reference methods. Of 31 findings by LC/TOF-MS, 27 could be confirmed by the reference methods, and only one of the unconfirmed findings was an apparent false positive. Quantification without primary reference substances was performed by LC equipped with a chemiluminescence nitrogen detector (CLND), using the detector's equimolar response to nitrogen. The quantitative results were consistent with those by the reference methods, the mean relative difference between results being only 11%. Analysis was rapid and simple, and sample preparation comprised only dilution. We conclude that the combination of LC/TOF-MS and LC/CLND provides an ideal solution for the analysis of drugs in seized material, without an inevitable need for primary reference substances. Figure 3 shows a simple total ion chromatogram of a seized sample, revealing 2,5-dimethoxy-4-iodophenethylamine (2CI), a designer amphetamine.


Figure 3: Total ion chromatogram of a seized street drug sample containing 2,5-dimethoxy-4-iodophenethylamine, a designer amphetamine. Compared with biological samples, the matrix is relatively simple and identification is unambiguous and straightforward even without a primary reference standard.
Utilization of Isotope Patterns
While several new generation LC/TOF-MS instruments allow mass determination with accuracy better than 5 ppm and resolution of 5000–10000 (FWHM), a major step forward was combining this capability with the full utilization of isotopic patterns. Formerly, only a rough characterization of isotopic patterns could be performed by LC/TOF-MS. Because of a novel design of ion transfer optics and detector technology, TOF-MS is able to measure the isotopic ratios within a 2% intensity precision. A new algorithm called the SigmaFit (Bruker Daltonik GmbH) indicates the exact numerical match of theoretical and measured isotopic patterns, providing an interesting, still unexplored parameter to aid identification in broad-scale screening. It gives an even better criterion for differentiation than accurate mass, these two parameters together allowing unambiguous identification of small molecules.29 Additional techniques, however, such as in-source CID, are needed for the differentiation of structural isomers with identical elemental formulae. Figure 4 displays the strong identification power of a combination of accurate mass and the SigmaFit over a wide dynamic range. The lower the sigma value (usually < 0.05), the better the isotopic match.

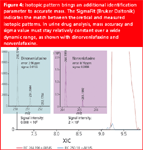
Figure 4: Isotopic pattern brings an additional identification parameter to accurate mass. The SigmaFit (Bruker Daltonik) indicates the match between theoretical and measured isotopic patterns. In urine drug analysis, mass accuracy and sigma value must stay relatively constant over a wide dynamic range, as shown with dinorvenlafaxine and norvenlafaxine.
Conclusions
LC/TOF-MS is a versatile and cost-effective analytical tool that is superior to many other MS techniques in terms of sensitivity, speed, mass accuracy and mass resolution. This is particularly true with the new generation bench-top instruments with improved performance; these may have 5 ppm mass accuracy and isotopic pattern matching over a wide dynamic range. High-throughput accurate mass measurement, however, requires appropriate data processing and reporting machinery, which is not necessarily an option with all brands. The screening approaches presented here have proven feasible in the fields of forensic science and toxicology, but they are equally applicable to other disciplines requiring comprehensive substance monitoring, such as environmental and food analysis and industrial hygiene.
Acknowledgements
The authors thank Anna Pelander, Matthias Pelzing, Merja Gergov and Erkki Vuori for collaboration.
Suvi Ojanperä is a postgraduate at the Department of Forensic Medicine, University of Helsinki. She researches developing methods for forensic drug analysis without reference standards. Ilkka Ojanperä is laboratory director at the same department. His research interests focus on broad-scale drug screening, drug-alcohol interactions and pharmacogenetics within forensic toxicology.
References
1. H.H. Maurer, J. Chromatogr. B, 733, 3–25 (1999).
2. O.H. Drummer, J. Chromatogr. B, 733, 27–45 (1999).
3. R.A. de Zeeuw, J. Chromatogr. B, 811, 3–12 (2004).
4. H.H. Maurer, Comb. Chem. High Throughput Screen., 3, 461–474 (2000).
5. H. Hoja et al., J. Anal. Toxicol., 21, 116–124 (1997).
6. H.H. Maurer, Clin. Chem. Lab. Med., 42, 1310–1324 (2004).
7. W. Weinmann et al., J. Am. Soc. Mass Spectrom., 10, 1028–1037 (1999).
8. M. Rittner et al., J. Anal. Toxicol., 25, 115–124 (2001).
9. F. Saint-Marcoux, G. Lachâtre and P. Marquet, J. Am. Soc. Mass Spectrom., 14, 14–22 (2003).
10. M.J. Bogusz et al., J. Chromatogr. A, 844, 409–418 (1999).
11. W. Weinmann, M. Gergov and M. Goerner, Analusis, 28, 934–941 (2000).
12. P. Marquet et al., J. Chromatogr. B, 789, 9–18 (2003).
13. T.N. Decaestecker et al., Anal. Chem., 76, 6365–6373 (2004).
14. M. Gergov et al., Rapid Commun. Mass Spectrom., 18, 1039–1046 (2004).
15. M. Gergov, I. Ojanperä and E. Vuori, J. Chromatogr. B, 795, 41–53 (2003).
16. R. Dams et al., J. Am. Soc. Mass Spectrom., 14, 1290–1294 (2003).
17. B.J. Kimble et al., J. Chromatogr. Sci., 12, 647–655 (1974).
18. A.S. Fang et al., Comb. Chem. High Throughput Screen., 1, 23–33 (1998).
19. L. Fang et al., Rapid Commun. Mass Spectrom., 16, 1440–1447 (2002).
20. M.P. Balogh, LCGC Europe, 17(3), 152–159 (2004).
21. H. Zhang et al., Anal. Chem., 72, 3342–3348 (2000).
22. M. Maizels and W.L. Budde, Anal. Chem., 73, 5436–5440 (2001).
23. M.W.F. Nielen et al., Rapid Commun. Mass Spectrom., 15, 1577–1585 (2001).
24. M. Gergov et al., Rapid Commun. Mass Spectrom., 15, 521–526 (2001).
25. A. Pelander et al., Anal. Chem., 75, 5710–5718 (2003).
26. I. Ojanperä et al., J. Anal. Toxicol., 29, 34–40 (2005).
27. K. Pfleger, H.H. Maurer and A. Weber, eds., Mass Spectral and GC Data of Drugs Poisons, Pesticides, Pollutants and Their metabolites, 2nd ed, (VCH, Weinheim, Germany, 1992), 423.
28. S. Laks et al., Anal. Chem., 76, 7375–7379 (2004).
29. S. Laks et al., 21st Montreux LC–MS Symposium, Abstracts of Poster Presentations, 102 (2004).
















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.




