Extraction of Micropollutants From Size-Limited Solid Samples
LCGC Europe
The quantitative extraction and subsequent purification of trace contaminants from (semi-)solid environmental and food matrices of regular size (that is, a few grams) is still recognized as a challenging task, typically accomplished through relatively complex off-line multistep treatment procedures. When these conventional sample preparation procedures are applied to the treatment of size-limited samples (of less than 1 g), the difficulties increase. This review discusses the different analytical strategies that can be adopted to overcome (or at least reduce) these difficulties when chromatographic techniques are involved for final instrumental determination.


Photo Credit: Gunther Kleinert/ EyeEm/Getty Images

Lourdes Ramos1 and Bruce Richter2
1Department of Instrumental Analysis and Environmental Chemistry, IQOG-CISC, Madrid, Spain
2Agilent Technologies, Wilmington, Delaware, USA


The quantitative extraction and subsequent purification of trace contaminants from (semi-)solid environmental and food matrices of regular size (that is, a few grams) is still recognized as a challenging task, typically accomplished through relatively complex off-line multistep treatment procedures. When these conventional sample preparation procedures are applied to the treatment of size-limited samples (of less than 1 g), the difficulties increase. This review discusses the different analytical strategies that can be adopted to overcome (or at least reduce) these difficulties when chromatographic techniques are involved for final instrumental determination.
The analysis of minor organic components in solid and semisolid environmental samples and foodstuffs is typically regarded as a challenging task. This consideration derives from the usually complex and heterogeneous nature of these matrices and the variety of mechanisms governing the retention of trace analytes on them, which range from adsorption or absorption to integration of the target compound into the matrix structure. Whatever the strength of the retention, accurate determination of residual organic analytes by direct sample analysis is in most cases virtually impossible, making some level of sample preparation before instrumental compound determination mandatory. The required level of sample pretreatment (understood here as analytical operations performed for analyte extraction, purification, and concentration) will depend on the selectivity and sensitivity offered by the technique selected for final separation and detection of the analyte and, indeed, on the criteria of the analyst. Nowadays, the analytical determination of residues of legacy and emerging organic micropollutants of medium and high volatility is usually accomplished by gas chromatography (GC) coupled to highly selective and sensitive detectors. Although detectors, including microelectron capture detectors (µECDs), nitrogen or phosphorous detectors (NDs or PDs), and many others, are still used in many control laboratories, the benchmark for these determinations lies with the modern mass spectrometry (MS)-based detectors. These instruments allow the simultaneous accurate determination of a significantly large number of pollutants in relatively complex extracts within a single chromatographic run. The enhanced selectivity offered by the modern hyphenated and hybrid MS detectors improves the signal-to-noise (S/N) ratios and reduces interferences. The former ultimately results in lower limits of detection (LODs) and allows accurate analyte determination at concentration levels much lower than expected only a decade ago.
Conventional sample preparation protocols used for the analysis of trace organic pollutants in (semi-)solid matrices involve the exhaustive (and, in most instances, nonselective) extraction of the target compounds from the matrix structure. This is followed by the thorough purification of the obtained complex extract and the final concentration of the analytes of interest before instrumental analysis. In general, integration among these several treatment steps, and particularly their automation, is very limited. This means that the methods to be considered are limited in terms of analysis response and sample throughput, as well as prone to analyte loss or contamination because of the constant manual manipulation of the sample and extracts. The enhanced detectability provided by modern GC–MS systems used for these types of determinations means that, in most instances, only a small fraction of, typically, 1–2 µL out of the final 500–1000âµL concentrated extract, is used for analyte instrumental determination. In principle, this means that accurate analyte detection and quantification should be possible, even if the initial amount of sample is reduced from the typical 2–20 g to a representative-size sample of a few grams (0.5–5 g). In addition, the volume of the final purified extract can be proportionally reduced (for example, to 150–300 µL) and large-volume injection (LVI) can be used to ensure that a similar amount of analyte to that involved in the conventional large-scale procedure is injected into the instrument.
This principle is the base for a plethora of widely accepted miniaturized analytical techniques developed during the last two decades for the treatment of liquid and viscous samples. Some of these techniques, such as miniaturized on-line solidâphase extraction (SPE), solid-phase microextraction (SPME), and stir-bar sorptive extraction (SBSE), have been implemented in control and routine laboratories and have demonstrated their performance for the analysis of both major and trace components. Other liquid-phase–based microextraction techniques, in particular hollow fibreâprotected solvent microextraction, in both its two or three-phase formats, and dispersive liquid–liquid microextraction (DLLME), are rapidly increasing in popularity and practical application range. In contrast, the application of such an approach for the treatment of (semi-)solid samples has been much more limited in the specialized literature, where in many instances it has been limited to the academic demonstration of the proof of concept, and it is essentially nonexistent in commercial laboratories. Several reasons are responsible for this different level of development and acceptance, and the most relevant are identified and discussed in this article. Whatever the reasons, the most immediate consequence is that, even today, sample preparation procedures used in most laboratories for the treatment of (semi-)solid samples are well established and validated protocols that are applied as described for conventional-size samples, irrespective of the actual initial sample amount. In our opinion, this approach is probably far from being the best option from both a practical and an analytical point of view, and the development of more size-suited procedures would be preferable.
This article discusses the advantages and shortcomings of miniaturization for the treatment of size-limited solid and (semi-)solid samples. The practicality of this approach will be discussed on the base of representative application examples dealing with the analysis of minor (that is, trace) organic pollutants because of the greater difficulty typically associated with this type of determination. The focus will be on those pollutants with medium and high volatility that will involve GC-based systems for instrumental determination, although gaseous compounds will not be considered because their extraction is typically accomplished using different extraction techniques. Nevertheless, if relevant, examples involving other types of separation techniques, such as liquid chromatography (LC), will also be considered. In all cases, applications and techniques dealing with the analysis of real-life (that is, nonspiked) samples will be preferred.

Miniaturized Sample Preparation: Advantages and Limitations
As previously indicated, the miniaturized treatment of liquid and viscous (including slurry-like) matrices can now be considered an achieved goal. The widespread use and implementation of a number of these well-accepted and established extraction techniques in both research and control laboratories supports this statement. The success of techniques such as SPME, on-line SPE, SBSE, and the more recently introduced DLLME or disposable pipette extraction (DPX, a miniaturized version of dispersive-SPE), relies, at least partially, on the several advantages derived from their miniaturized nature. Most of these techniques allow a significant reduction (and in some cases, virtual elimination) of the use of organic solvents and reagents compared with the corresponding large-scale counterparts. In many instances, they also allow the completion of the sample preparation protocol within a shorter time frame by significantly reducing (or avoiding) the need for a concentration step in between treatment steps. The elimination of this need has also facilitated the hyphenation and integration among the several treatments and techniques involved in the analytical process. Hyphenated or not, the miniaturization of the analytical processes has been recognized as a mandatory step when trying to set up (at least partially) integrated and (semi-)automated systems contributing to increased sample throughput and minimized waste generation and energy consumption. Moreover, these methods have demonstrated equally satisfactory analytical results to conventional (that is, large-scale) methods, even for complex determinations, such as the analysis of trace micropollutants in influents of wastewater treatment plants and river waters (1).
Equivalent application examples described in the literature for the miniaturized treatment of solid samples reported similar benefits to those stated from liquid matrices, viz. reduced solvent and reagents consumption, improved capability of coupling between the extraction step and the subsequent treatments, the possibility to yield readyâforâanalysis extracts, less waste generation, and improved analytical response as a result of the shortened analytical time (2). As for liquid samples, experimental results have demonstrated that the reported miniaturized protocols can achieve equally satisfactory analytical performances to their equivalent large-scale protocols, even for difficult determinations, such as residues of polyaromatic hydrocarbons (PAHs) in sediments, polychlorinated biphenyls (PCBs) in fatty animal tissues, or pesticides in single insects (see Table 1).
Despite the many positive features and advantages shared by the miniaturized treatment of liquid and solid samples, the sharp differences between the levels of development and general acceptance reached by liquid samples compared to solid samples makes it clear that significant differences exist between these two application areas. Several aspects can be considered in this respect, but probably the most relevant would be: (i) the natural reticence of researchers to work with small heterogeneous samples, and (ii) the lack of appropriate commercial miniaturized instrumentation required to implement some of these methods. Regarding the former aspect, we should accept that the heterogeneity of some (semi-)solid matrices can make the collection of a representative subsample difficult. In such cases, we tend to assume that the larger the subsample analyzed, the more representative the analytical result. However, that is not necessarily the case. The representativeness of the subsample analyzed can only be guaranteed by the application of appropriate sampling protocols, which is out of the scope of this discussion. Whatever the sampling protocol applied, and assuming that a representative subsample is available for analysis, protocols for the treatment of (semi-)solid environmental and foodstuffs need to include a number of sample pretreatments prior to analyte extraction. In the case of trace pollutants, these pretreatment operations must include agglomerates disaggregation, sample chopping and mixing, water and humidity removal (for example, by freeze drying in the case of foodstuffs), grinding of the dried sample, and, in some cases, sieving to an appropriate sample size particle (for example, in the case of soils and sediments). The aim is to increase the homogeneity of the sample subjected to analysis and to ensure proper contact among the matrix components and the extraction solvents and reagents during the extraction step. When done properly, the enhanced homogeneity should also enable smaller fractions that are equally representative of the selected subsample to be defined. In other words, this approach should make the analysis of smaller fractions of the initial sample possible without compromising the representativeness of the final results. For obvious reasons, the final size of these smaller fractions is not expected to be the same for all matrices. The minimum amount of sample required for the analysis is determined by the heterogeneity of the original sample, the nature and effectiveness of the pretreatment operations performed, and the sensitivity provided by the instrumental step, all carefully determined by the analyst. Nevertheless, such considerations do not apply in size-limited samples, that is, in those in which the initial amount of sample available is determined by the sample nature (for example, the analysis of single insects) or the sampling procedure (biopsies). In these cases, direct application of the conventional methodologies results in an extremely high analyte dilution, which significantly increases the risk of analyte loss or contamination during sample treatment. Another undesirable consequence is the significant increase of the blank values when compared to those of the target analyte - something that can negatively affect the accuracy of the determination. In our opinion, for these types of samples, the analytical method should preferably be adapted to the sample characteristics, which makes miniaturization one of the better - if not the best -analytical alternatives.
A second identifiable aspect hampering progress in miniaturized preparation of solid matrices is the limited commercial miniaturized instrumentation adapted for such methods and, in particular, for sample extraction. While some of the miniaturized techniques used for the treatment of liquids can be applied for analyte purification, the number of instruments really adapted for the extraction of small-size solid samples is rather limited. In this context, strategies for the in-house scaling-down of the methods and the setting-up of homemade size-adapted instruments have been adopted with satisfactory results. The advantages and shortcomings of these different analytical strategies will be discussed next through illustrative application studies.
Analytical Techniques for the Extraction of Size-Limited Samples
When considering what techniques or strategies to use for size-limited samples, one has to take into account what techniques are easily scaled down in the laboratory from methods that are used for samples without size limitations, as well as what technologies are commercially available that can be used. Table 1 shows an overview of selected applications involving a variety of miniaturized methods used for the treatment of size-limited solid samples.
Manual Methods that Can be Scaled Down: Soxhlet extraction has been used for many years, and is often used for large samples with corresponding copious amounts of solvents because this configuration is so common. It has been reported that Soxhlet was used without any modification for the treatment of small-size samples (11), despite the final analyte dilution. However, one can purchase much smaller glassware to perform Soxhlet extractions on samples of less than a gram in size (3). With smaller samples, the long times typically required for extractions can be shortened. As for any other method development and validation, optimization should preferably be performed using standard reference materials to ensure that the correct solvent and time have been used for the target analytes and matrix.
Another more traditional technique for the extraction of solid samples, solid–liquid extraction (SLE), can be easily miniaturized and used for small samples. Glass vials can be loaded with the weighed samples and the appropriate amount of extraction solvent and then shaken either by hand or a mechanical type of shaker to ensure good interaction of the sample and solvent. The samples can also be heated on hot plates or water baths to temperatures above ambient but below the boiling point of the extraction solvent to speed up the extraction process. The nonextracted sample can be separated from the liquid by either centrifugation or filtration to obtain the liquid extract for subsequent sample processing prior to analysis. Despite the difficulty of automating the technique because of the latter steps (19), SLE is still a common technique used in laboratories for its simplicity. In recent years, its use in combination with alternative, green and more selective solvents, such as ionic liquids (ILs) and deep eutectic solvents (DES), has helped to solve additional shortcomings associated with its use with conventional volatile organic solvents, such as limited selectivity. As a typical example, Helalat-Nezhad et al. (4) reported on the feasibility of choline chloride-oxalic (ChClâOx) to completely dissolve marine biological samples (fish and macroalgae samples). The dissolved extract (5 mL) was then percolated through a SiO2 column for selective retention of biogenic material, while the targeted PAHs were quantitatively eluted with 5 mL of cyclohexane. The authors reported recoveries in the 72–110% range, and LODs of 0.50–3.08 ng/g using
LC–fluorescence (FL) detection and only 100 mg of sample for analysis. Interestingly, the extraction was completed at 55 ºC in 30 min at atmospheric pressure, which is an extraction time in the range of those used with other enhanced extraction techniques, despite the less drastic extraction conditions.
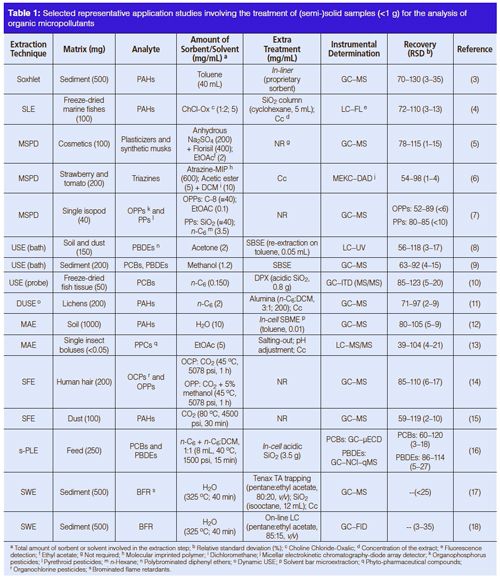
Matrix solid-phase dispersion (MSPD) is a technique that is often used for solid samples of more conventional sizes. In this technique, the solid or semisolid sample is mixed with an appropriate solid-phase sorbent to disperse the sample. This dispersed sample is then loaded into an SPE-type cartridge and subsequently treated like an SPE cartridge. The sorbent in the cartridge is eluted with the appropriate organic solvent to obtain a liquid extract containing the target analytes and essentially free of (nonstructurally related) interferences. This process can be easily miniaturized by adjusting the amount of solid sorbent and the volume of the cartridge used. The addition of a cosorbent on the base of the MSPD column to retain coeluted (structurally related) matrix components (5) or the use of highly selective sorbents, like molecular imprinted polymers (MIPs) (6), can result in readyâfor-analysis extracts, thereby minimizing extract manipulation and improving analytical response times. Despite its many positive features, MSPD is difficult to automate and it can easily fail at efficiently extracting analytes from various classes at the same time, except if a careful optimization of the extraction conditions is performed (7,13) (Figure 1).

Ultrasonic extraction (USE) is another technique frequently used for conventionally sized samples (10–30 g) that can be scaled down to work with size-limited samples. USE is typically performed one of two ways: either extraction in an ultrasonic bath or extraction with an ultrasonic tipped probe. To perform USE in a bath, the solid sample is placed in an appropriately sized glass vial to contain the sample and solvent and then placed in the bath for a preselected extraction time (8,9). Afterwards, the solid and solvent are separated using either centrifugation or filtration. In the dynamic mode, the sample is typically placed in a refillable column, which is immersed in the ultrasonic bath. The extraction solvent flows continuously through the sample and is transferred either to a vial (11) or on-line to the instrumental analysis (21). If a sonic probe is used, it is placed directly in the vial containing the sample and extraction solvent. This direct contact between the tip and the sample results in a very efficient extraction and shortened extraction times (that is, a few minutes or even seconds suffice for matrix disaggregation and analyte extraction) (10). However, because of the large amount of heat dissipation, the sample often needs to be cooled, and volatile and labile analytes can easily be lost. Whatever the USE format selected, subsequent concentration and purification of the extracts before separation+detection is most frequently required because of the nonselective extraction process. A variety of techniques ranging from classical open columns (11) to disposable pipettes (10) or stir bars followed by analyte back extraction (when combined with LC [8]) or thermal desorption (with GC [9]) have been used. Approaches involving on-line systems are based on dynamic USE and are scarce in the literature (21).
Methods with Automation that Can be Scaled Down: The discussed methods can be easily adapted to small samples, but they are, in general, manual and can involve a lot of hands-on manipulation. While this may be fine for a few samples, for laboratories that have high sample loads, methods that use at least some automation would be advantageous.
Microwave assisted extraction (MAE) is often used for the extraction of environmental and food-related samples of conventional sizes. One advantage of MAE is the rapid heating caused by the microwave radiation. Some solvents do not absorb much energy and the vessel that contains the sample is constructed of materials that absorb the microwave energy and cause the sample to heat up. Alternatively, the extraction solvent can be mixed with a polar solvent. However, this can negatively affect the efficiency of the extraction process (13). In general terms, MAE can be performed in two basic ways: open vessel or closed vessel. Open vessel MAE is not performed as often as closed vessel because organic solvents can be heated to temperatures above their boiling points very quickly, and that could cause sample loss. There have been some reports of using focused microwaves along with Soxhlet-like glassware for open vessel MAE so that the boiling solvent is captured via the condenser (22). However, to the best of our knowledge, the technique has not been applied for the analysis of trace organic pollutants in small-size samples. The use of closed vessels appears to be the more common approach used for MAE. An advantage of this approach is that the use of closed vessels allows extractions to take place above the boiling point of the solvents chosen. Operation at these elevated temperatures and pressures (50–150 ºC; 100–500 psi) facilitates the rapid extraction of solid samples and minimizes the amount of solvent used. In addition, multiple samples can be extracted simultaneously in single batches to improve the overall productivity (up to 50 in a batch). Different vessel sizes are available from commercial vendors to accommodate smaller sample sizes (1 g or less). There are also systems that can extract one sample at a time and are equipped with autosampler-type robotics that accommodate many samples for unattended operation. After the extraction, the sample vessels are allowed to cool and the contents are filtered to separate the extracted solid sample from the solvent containing the extracted compounds. Depending on the complexity of the sample, subsequent (off-line) cleanup of the obtained extracts before instrumental analysis can become mandatory, making the entire sample preparation process more labour-intensive and time-consuming. Different analytical strategies have been explored in the literature to solve this type of shortcoming and to improve the selectivity of the extraction step. The development by Guo and Lee was an imaginative and interesting example (12). They proposed the combined use of MAE for the extraction of PAHs from soil with in-cell solvent bar microextraction (SBME) for simultaneous purification and selective preconcentration of the target analytes. In this study, 1 g of soil was extracted for 20 min using water at 60 ºC as a green and inexpensive solvent. The minimum volume of solvent recommended by the instrument manufacturer - 10 mL - was enough to immerse the sample completely and to ensure that the solvent bar could move and tumble freely into the extraction vessel to promote appropriate analyte enrichment during the extraction step. The results obtained by MAE–SBME were similar to or slightly better than those obtained with conventional MAE followed by analyte purification with SPE. However, in the former case, analyte extraction, purification, and preconcentration were accomplished in a single step. This application example clearly illustrates the potential of innovative approaches, such as MAE–SBME, for the simple, fast, and efficient preparation of complex matrices.
Supercritical fluid extraction (SFE) has been used for the extraction of solid samples for several decades. In SFE, carbon dioxide, with added organic modifiers like methanol, is used as the extraction solvent. The extraction takes place at elevated temperatures and pressures (35–100 ºC; 2000–10,000 psi). The solvent (supercritical fluid) flows through the extraction cell containing the solid sample. As with MAE, these conditions facilitate rapid extractions, and small volumes of organic solvents are consumed, especially with small samples. SFE can be used in two basic configurations for the preparation of size-limited samples: off-line and on-line. In off-line SFE, the system is a standâalone instrument that can extract samples individually or in parallel. The extracts are collected in organic solvents directly or on solid supports that are subsequently washed with organic solvents. These extracts can then be analyzed via separate chromatographic instrumentation. In on-line SFE, the effluent from the extraction vessel is directed to an analyte trap or concentrator column. Once the extraction has been completed, the flow path is changed via a switching valve, so that the trap or concentrator column is in-line with the chromatographic separation column. Instrumental analysis is then performed on the extracted and collected sample. Either of these configurations can be adapted to accommodate small samples for extraction, as demonstrated by Cuong et al. (14) in a recent example dealing with the SFE of pesticides of varying polarity from hair collected from nonexposed humans. The optimized method allowed SFE of nonpolar organochlorine pesticides (OCPs), such as hexachlorobenzene, lindane, butapon, p,p′-DDE, and p,p′-DDT, with pure CO2 (recoveries in the 51–92% range; standard deviation [SD], 7–17). However, the quantitative SFE of more polar organophosphorus pesticides (OPPs), such as dichlorvos, 2,4-D, and chlorofos, required modification of supercritical CO2 with 5% methanol (recoveries, 71–81%; SD, 7–20). When combined with a brief static SFE step for previous selective removal of endogenous material, the proposed dynamic SFE method resulted in clean chromatograms and allowed accurate analyte determination at the µg/g level.
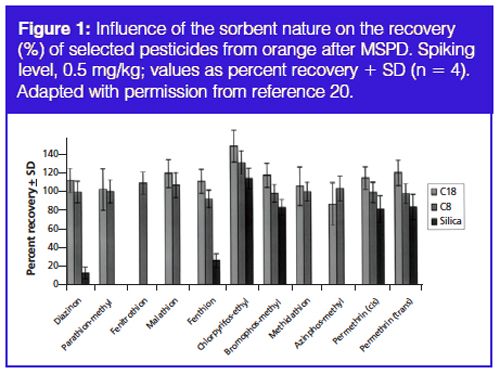
Pressurized liquid extraction (PLE) is also known as accelerated solvent extraction (ASE) and pressurized solvent extraction (PSE). PLE is similar to SFE, except organic solvents or solvent mixtures are used as the extraction fluid, and no carbon dioxide is used in the extraction. Elevated temperatures and pressures are used (40–200 ºC and 1500–2000 psi) to facilitate rapid extractions with small solvent volumes. The solvent flows through the extraction cell containing the solid sample, and the extracts are collected in glass vials or bottles. There are some unique advantages to SFE and PLE as compared to MAE because of the flow through mode of operation. The sample extracts are filtered as they are collected because the extraction cells contain fritted materials in SFE and PLE. With the flow through operation, sorbents can be placed in the flow path and unwanted interferences can be trapped as the samples are extracted. Sample extracts that are filtered and contain fewer interferences can therefore be easily produced with SFE and PLE. When the sorbents are packed in the PLE extraction cell, the technique is called selective-PLE (s-PLE). This latter arrangement (see Figure 2 for a typical example for the analysis of persistent organic pollutants [POPs] in feedstuffs) has been shown to contribute to minimized sample manipulation, sorbent, and solvent consumption, and to significantly shorten the analytical times (16).

While PLE is often performed on larger samples (5–100 g), commercial instrumentation is available with extraction cells as small as 1 mL. These will work for small samples (1 g or less), but the results might not be as satisfactory as with laboratory or in-house built instruments because of dead volume and other considerations (3,16). More importantly, this type of miniaturized system is now the only feasible method for on-line coupling between PLE and the instrumental analytical step (23).
Dynamic PLE has generally been adopted in on-line coupling of subcritical water extraction (SWE, a particular type of PLE) with subsequent analyte separation+detection typically done by LC (18). Unlike conventional PLE, green water-based solvents are used for SWE. The technique also involves much higher extraction temperatures (typically, 100–300 ºC), making subsequent depressurization and cooling down of the cell eluent mandatory before analyte trapping. These extra elements complicate the setting up of SWE devices and could be responsible for the limited progress observed in this field, despite its remarkable analytical features.
Desirable Future Developments and Conclusion
As previously stated, there have been many reports in the literature on the use of laboratory-built instruments either directly coupled to GC–MS or LC–MS or in a stand-alone configuration for the extraction and cleanup of sizeâlimited solid and semisolid samples. These are useful for sizeâlimited samples across a broad spectrum of matrices and analyte types. However, there are few commercially available instruments with these capabilities. In this sense, probably the most significant addition is the recent introduction of an SFE–SFC system that offers extraction vessels as small as 0.2- and 0.5-mL, and is ideally suitable for the extraction of milligram quantities of solid samples followed by the determination of target analytes by SFC–UV or SFC–MS.
We are not aware of other commercially available instruments. While small volume PLE has been successfully demonstrated in the literature (3,16,23), the smallest volume of commercial extraction cells available for this technology is 1 mL, which can still be too big for certain applications. The same can be said of the other techniques covered in previous sections. The authors encourage commercial vendors to carefully consider the development and commercialization of sample preparation and extraction instrumentation that has been optimized for use with samples of less than 1 g in size. This is important as concern for the environment grows alongside the drive for more efficient and more “green” laboratory practices.
References
- M. Kock-Schulmeyer, M. Villagrasa, M. Lopez de Alda, R. Cespedes-Sanchez, F. Ventura, and D. Barcelo, Sci. Tot. Environ.458, 466–476 (2013).
- J. Escobar and L. Ramos, Trends Anal. Chem. 71, 275–281 (2015).
- L. Ramos, J.J. Vreuls, and U.A.T. Brinkman, J. Chromatogr. A891, 275–286 (2000).
- Z. Helalat-Nezhad, K. Ghanemi, and M. Fallah-Mehrjardi, J. Chromatogr. A1394, 46–53 (2015).
- M. Llompart, M. Celeiro, J.P. Lamas, L. Sanchez-Prado, M. Lores, and C. Garcia-Jares, J. Chromatogr. A1293, 10–19 (2013).
- Y. Wen, L. Chen, J. Li, Y. Ma, S. Xu, Z. Zhang, Z. Niu, and J. Chho, Electrophoresis 33, 2454–2463 (2012).
- E.M. Kristenson, S. Shahmiri, C.J. Slooten, J.J. Vreuls, and U.A.T. Brinkman, Chromatographia59, 315–320 (2004).
- C. Yu and B. Hu, J. Chromatogr. A1160, 71–80 (2007).
- P. Tolgyessy, B. Vrana, and K. Silharova, Chromatographia76, 177–185 (2013).
- M. Pena-Abaurrea, V.S. Garcia de la Torre, and L. Ramos, J. Chromatogr. A1317, 223–229 (2013).
- C. Domeño, M. Blasco, C. Sánchez, and C. Nerín, Anal. Chim. Acta569, 103–112 (2006).
- L. Guo and H.K. Lee, J. Chromatogr. A1286, 9–15 (2013).
- L. Haroune, R. Cassoulet, M.P. Lafontaine, M. Belisle, D. Garant, F. Pelletier, H. Cabana, and J.P. Bellenger, Anal. Chim. Acta 891, 160–170 (2015).
- L.P. Cuong, M.I. Evgen’ev, and F.M. Gumerov, J. Supercrit. Fluid.61, 86–91 (2012).
- Y. Ren, J.J. Lian, H.X. Xue, J.M. Chen, and T.T. Cheng, Ann. Chim.96, 669–680 (2006).
- M. Pena-Abaurrea, J.J. Ramos, M.J. Gonzalez, and L. Ramos, J. Chromatogr. A1273, 18–25 (2013).
- T. Hyotylainen, K. Hartonen, S. Syndjoki, and M.L. Riekkola, Chromatographia53, 301–305 (2001).
- K. Kuosmanen, T. Hyötyläinen, K. Hartonen, and M.L. Riekkola, J. Chromatogr. A943, 113–122 (2002).
- L. Ramos, J.J. Vreuls, and U.A.T. Brinkman, Environ. Sci. Technol. 33, 3254–3259 (1999).
- E.M. Kristenson, E.G.J. Haverkate, C.J. Slooten, L. Ramos, R.J.J. Vreuls, and U.A.Th. Brinkman, J. Chromatogr. A917(1–2), 277–286 (2001).
- C. Sanchez, M. Ericsson, H. Carlsson, A. Colmsjö, and E. Dyremark, J. Chromatogr. A957, 227–234 (2002).
- F. Preigo-Capte and M.G. Luque de Castr, Talanta65, 98–103 (2005).
- Y.S. Zhou, W.M. Liu, J.H. Zhao, and Y.F. Guan, Chin. J. Anal. Chem.33, 1231–1234 (2005).
Lourdes Ramos is currently a Senior Scientific Researcher of the Scientific Research Council (CSIC, Madrid) of Spain at the Department of Instrumental Analysis and Environmental Chemistry, in the Institute of Organic Chemistry. Her research interests include the development of novel sample preparation methods for the determination of trace pollutants in environmental and food samples (including their scaling down and miniaturization) and the evaluation of new chromatographic techniques, especially GC×GC-based approaches, for unravelling the composition of complex mixtures of organic microcontaminants.
Bruce Richter is currently an R&D Manager in the Agilent CrossLabs Group (ACG) of Agilent Technologies in Wilmington, Delaware, USA. He has been involved in the development of sample preparation technologies for many years, and he was a member of the R&D team that developed accelerated solvent extraction (ASE) at Dionex Corporation (now Thermo Scientific). His research interests include LC column development, GC column development, and the development of new approaches for improving overall analytical performance through innovative sample preparation technologies.







.jpg")












