Evaluation of Fatty Acids as Biochemical Markers for Source Identification of Indian Opium
LCGC Europe
This method describes an attempt to identify the source of licit Indian opium based on the fatty acid profile. The analysis was based on gas chromatography (GC) with flame ionization (FID) and mass spectrometric detectors (MS). A total of 124 Indian opium samples were collected and fingerprinted for the presence of various fatty acids. Qualitative analysis of fatty acids indicated the acids such as behenic, stearic and lignoceric were significant biochemical markers, making it a useful method to identify the source of opium for forensic purposes.
Opium is partly dried latex obtained from the unripe capsules of Papaver somniferum L. It is cultivated mainly in the Indian subcontinent, Turkey, China, France, Spain and Australia. India is the major licit producer and a principal supplier of opium to the world market for medicinal purposes. About 20000 hectares of land has been used in the specified areas of Madhya Pradesh (MP), Uttar Pradesh (UP) and Rajasthan (Raj) of India for the cultivation of opium in India under the supervision of the Central Bureau of Narcotics, Government of India.1



However, opium is a narcotic commodity and its illicit cultivation, production, manufacturing, possession, sales, purchase and trafficking attracts stringent punishment under the Narcotic Drugs and Psychotropic Substance (NDPS) Act 1985, Government of India. The sentence is Rupees 200000, equivalent to a US $4200 fine and 10–20 years imprisonment, which may go up to death penalty if the offence is repeated.
Opium ranks as the second most important item of globally seized commodity after cannabis (Cannabis sativa L). The worldwide report brought out by the Office of National Drug Control Policy, USA indicates that there was an increase of 152% in the production of illicit opium during the period 1985–1992.2 According to the United Nations Office on Drugs and Crime report (2003), the illicit opium production in the world was estimated to be 4000–6000 metric tonnes a year.3
Characterization of seized narcotic samples through laboratory analysis has long been recognized as a valuable tool to the law enforcement agencies in controlling the clandestine opium production and its illicit trafficking. Such characterization not only provides sufficient proof of identity for judiciary, but may also assist in establishing the trafficking patterns and eventually the source of the illicit opium.
Attempts have been made to find out the source of opium by analysing the various chemical constituents including fatty acids in opium to help the law enforcement agencies for checking its illicit production and trafficking under the United Nations opium characterization programme.4
Fatty acids are one of the important constituents in opium latex. Earlier, profiling of fatty acids was reported under the United Nations Opium Research Programme.5 Around 32 samples from countries, such as Yugoslavia, Greece, Turkey, Iran, Afghanistan, Pakistan, India, Vietnam, Laos, China, Korea, Japan, Ecuador and Mexico were analysed for fatty acid profile using gas chromatography coupled with thermistor detector to pinpoint the geographical origin of opium samples by means of the fatty acids.5 There were certain limitations in the reported method, which include
- low resolution of fatty acids
- a time-consuming extraction procedure
- low sensitivity with high noise level
- samples analysed from India were too meager (five samples).
The small sample size of Indian opium may not permit a definite conclusion for source identification. There is no detailed information on the fatty acid profile of Indian opium samples.
Furthermore, the use of fatty acid profile as a possible biochemical marker for the discrimination of the licit opium growing divisions of India has not been explored. Hence, it was considered worthwhile to study the fatty acid profile of opium samples originating from licit opium growing divisions of India in order to evaluate the fatty acids, as possible biochemical descriptors for the source identification of Indian opium.
In recent years, GC coupled to flame ionization detection (FID) and mass spectrometry (MS) are now extensively used for the compositional determination of fatty acids in various plant and food materials.6,7 Short chain fatty acids (C2–C6) can be analysed without derivatization but many fatty acids (>C6) must be derivatized before analysis because of their low volatility or poor thermal stability.
The International Union of Pure and Applied Chemistry (IUPAC) reports standard methodology for this technique. It involves the alkaline sopanification of the extracted fat to break down the glycerides, the liberated fatty acids being esterified in the presence of methanol and boron trifluoride. Fatty acid methyl esters (FAMEs) are then extracted with an organic solvent and analysed using GC with flame ionization and mass spectrometry detectors. This technique is a useful tool for fingerprinting the fatty acids for the classification and differentiation of opium samples by comparing peaks and peak heights.
In the present study, a method was developed using gas chromatography with flame ionization detection, which is rapid, sensitive and accurate for the determination of fatty acids in Indian opium and unequivocal confirmation of these fatty acids by mass spectrometry. Furthermore, fatty acids were evaluated as possible biochemical markers for the source identification of Indian opium by the application of chemometrics.
Materials and Methods
Chemicals and reagents: All chemicals and reagents used were of analytical grade. Sodium methoxide, BF3–methanol, methanol, n-hexane, sodium sulphate were procured from E-Merck Limited, Mumbai, India. A mixture of standard FAMEs with a concentration of 1 mg/mL in dry dichloromethane was procured from Supelco, USA. Authentic samples of opium (n = 124) were collected in a semi-dried state from the states of MP, UP and Raj covering 14 divisions of licit opium growing divisions of India. Government opium and alkaloid works situated at Ghazipur, UP and Neemuch, MP facilitated the sample collection. The number of samples collected from each division were namely Garoth (MP, 3 samples), Jaora (MP, 3 samples), Mandsaur (MP, 18 samples), Neemuch (MP, 12 samples), Ujjain (MP, 7 samples), Bhilwara (Raj, 8 samples), Chittorgarh (Raj, 15 samples), Jhalwar (Raj, 9 samples), Kota (Raj, 17 samples), Pratapgarh (Raj, 6 samples), Barabanki (UP, 6 samples), Bareilly (UP, 6 samples), Faizabad (UP, 6 samples) and Tilhar (UP, 8 samples). The samples obtained from the above sources were dried at 105 °C for 2 h to a constant weight and powdered in a high-speed mechanical blender. Powdered samples were kept in a vacuum dessicator under anhydrous calcium chloride until further use. The values reported in the present study are on dry weight basis.
Preparation of standards: A mixture of thirty-seven standard FAMEs (1mg/mL each) was further diluted to a concentration of 100 mg/mL with dry dichloromethane.
Determination of the fat content by soxhlet extraction: One gram of powdered opium (dried to constant weight at 105 °C for 2 h) was extracted for six hours under reflux with hexanes by a soxhlet extractor. The extracted solvent was flash evaporated and the residue was dried at 100 °C for 30 min in an electric oven and percent fat (g%) in opium samples was calculated.
Preparation of fatty acid methyl esters (FAMEs): A method reported by Morrison and Smith was adopted for the preparation of FAMEs.9 The fat extracted by the soxhlet extractor was transferred to a polytetrafluoroethylene (PTFE) lined screw capped vial and 2 mL of 0.5 M sodium methoxide in methanol was added. The vial was kept in a boiling water bath (100 °C) for 10 min and cooled to room temperature. Then, 1–2 drops of BF3–methanol reagent was added and heated for 5 min in a water bath. The resultant mixture was cooled and dissolved in 1–2 mL of hexane, vortexed and kept for 30 min at room temperature for the hexane layer to separate out. An aliquot of the supernatant (hexane) layer was transferred into a Pasteur pipette (5 .75 inch) packed with anhydrous sodium sulphate (about 1 g) to remove water. After filtration, 1 µL of the filtrate was analysed by the gas chromatograph equipped with FID and MS detectors.
GC–MS conditions: All GC analyses were performed on a model Auto System XL GC gas chromatograph (PerkinElmer, USA) equipped with a split/splitless capillary injector and FID. Analytical separation was achieved on a capillary column (50% cyanopropylphenyl–50% methyl siloxane, PE–225, 30 m × 0.25 mm i.d., 0.25 µm film thickness; PerkinElmer, USA). The carrier gas (helium) flow-rate was kept as 1.1 mL/min.
The amount injected was 1 mL without split. Temperature settings were as follows: injector 230 °C; FID 230 °C. The oven temperature was held at 100 °C for 1 min and then programmed to 180 °C at 10 °C/min and held for 30 min at 180 °C. GC peak areas were integrated with Totalchrom Work Station software supplied along with the instrument (PerkinElmer, USA). The peak areas were normalized using unit response factors. The total amount of lauric (C12:0), myristic (C14:0), palmitic (C16:0), stearic (C18:0), oleic (C18:1), linoleic (C18:2), linolenic (C18:3), arachidic (C20:0), behenic (C22:0) and lignoceric (C24:0) acids was calculated as a hundredth of a percent.
All traces of fatty acids less than 0.1% were excluded in the study for the simple comparison. Samples were also analysed on a Turbo Mass GC–MS (PerkinElmer, USA) using the same column for MS identification of the GC components at electron impact (EI) ionization voltage 70 eV with a source temperature of 180 °C and an interface temperature of 250 °C. The GC retention time and mass fragmentation pattern of each component of opium was compared with that of an authentic compound for identification.
Statistical analysis: Statgraphics (version 5.0, Statistical Graphics Corporation, Maryland, USA) and Microcal origin software's (version 6.0, Microcal Software Incorporation, Northampton, USA) were used for chemometric (discriminant) analysis and plotting the discriminant scores.10
Results and Discussion
The percentage of fat in opium samples from different licit opium-growing divisions of India are shown in Table 1. The fat content of Indian opium samples showed wide variation in the range of 1.03–17 g%. However, the majority of Indian opium samples were found to have the fat content in the range of 8–12 g%. In the present method, lipid bound fatty acids and free fatty acids were derivatized directly from fat into their methyl esters using methanolic sodium methoxide and BF3 in methanol, which greatly reduces the time in the preparation of fatty acid methyl esters. In an earlier reported method,5 initially, unsopanifiable matter was removed from opium fat and then the fatty acids were derivatized into their methyl esters, which is found to be very laborious and time consuming.


Table 1: The fat content (g%) and amount (%area)* of fatty acids in the opium samples of licit opium growing divisions of India as determined by GC-FID
The usage of a capillary column instead of a packed column for the separation of FAMEs increased the resolution between the fatty acids (Figure 1). The peak identification was done by comparing the retention times of FAMEs in opium with that of standard FAMEs and also confirmed using a mass spectrometric detector by comparing the mass spectra of FAMEs in opium with that of standard FAMEs. Further, the peak was confirmed by matching the spectra of individual standards available from the National Institute of Standards and Technology (NIST, Gaithersburg, Maryland, USA) with that of the spectra of FAME in opium sample. Mass spectra of some of the FAMEs of opium are depicted in Figure 2. A total of ten fatty acids were detected and identified in all the opium samples analysed. The fatty acid methyl esters include lauric (C12:0), myristic (C14:0), palmitic (C16:0), stearic (C18:0), oleic (C18:1), linoleic (C18:2), linolenic (C18:3), arachidic (C20:0), behenic (C22:0) and lignoceric (C24:0). The relative standard deviation (RSD) of the five repetitive injections of fatty acids were observed to be 0.48%, 0.12%, 0.58%, 0.47%, 0.7%, 0.04%, 0.26%, 0.14%, 0.76%, 0.49% for retention time and 1.1%, 3.2%, 2.4%, 0.67%, 4.5%, 5.7%, 1.32%, 1.02%, 0.78%, 1.6% for peak area, respectively.
Each fatty acid was detected and quantified on the basis of area normalization method (% area). All the opium samples collected from licit opium-growing divisions of India were analysed using the above methodology for their fatty acid profile. Table 1 shows the mean percentage composition (% area) of fatty acids in the opium samples analysed. There was also a considerable variation in the relative composition of fatty acid in different opium samples. The fatty acids present in major quantity were linoleic, palmitic, oleic and myristic, while other fatty acids occurred in small quantities.

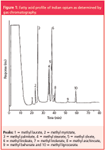
Figure 1: Fatty acid profile of Indian opium as determined by gas chromatography.
Qualitatively, significant results were observed in the fatty acid profile of licit opium growing divisions of India. Behenic acid was not detected in the divisions of Barabanki, Bareilly, Faizabad and Tilhar. Interestingly, all these divisions belonged to the state of Uttar Pradesh. Stearic acid was not detected in the divisions of Barabanki, Bareilly, Bhilwara, Faizabad and the majority of samples from Chittorgarh (10/15). Lignoceric acid was not detected in the divisions of Bareilly and Pratapgarh. The above observations clearly indicated that the samples originating from UP and Raj can be clearly differentiated from the divisions of MP by means of the fatty acids, namely behenic, stearic and lignoceric acids.


Figure 2: Comparison of the mass spectrum of standard FAMEs with that of FAMEs in opium sample. Spectra of (a) standard oleic acid methyl ester from the NIST library; (b) oleic acid methyl ester in Indian opium; (c) standard stearic acid methyl ester from the NIST library; (d) stearic acid methyl ester in Indian opium; (e) palmitic acid methyl ester from the NIST library; (f) palmitic acid methyl ester in Indian opium; (g) linolenic acid methyl ester from the NIST library; (h) linolenic acid methyl ester in Indian opium; (i) linoleic acid methyl ester from the NIST library; (j) linoleic acid methyl ester in Indian opium.
Chemometrics
In recent years chemometric techniques have been widely used for the classification and differentiation of opium samples of different origins. Several authors have published investigations applying different chemometric techniques in the differentiation and classification of opium samples according to their geographical origin and using different parameters.11,12 In the present study discriminant analysis of the fatty acid quantitative data was performed to find the optimal classifier based on fatty acid profile, which in turn minimizes classification errors.
A software-based multiple discriminant analysis (MDA) programme was used as a feature selection approach that provides better classification. Pictorial representation of multivariate data into a two-dimensional projection with minimum classification error was obtained by plotting the discriminant scores. Discriminant scores were derived and plotted as a graph (Figure 3).
The opium samples can be classified into three groups based on the fatty acid quantitative data (Figure 3). The samples from Mandsaur (14/18), Neemuch (8/12), Ujjain (4/7), Pratapgarh (4/6), Chittorgarh (5/15), Tilhar (1/8), Garoth (1/3), Jaora (1/3), Kota (3/17), Jhalwar (3/9) and Bhilwara (4/8) are falling within one group (group I). The samples from Barabanki (4/6), Chittorgarh (9/15), Faizabad (3/6), Bareilly (4/6), Tilhar (3/8), Mandsaur (2/18), Neemuch (2/12), Garoth (1/3), Jaora (2/3) and Jhalwar (2/9) are falling within the second group (group II). Finally, the samples from Kota (14/17), Jhalwar (3/9), Bhilwara (3/8) and Bareilly (1/6) constitute the third group (group III). The remaining samples (23/124) were found to be outside the classified groups and as well outside the two-dimensional projection.

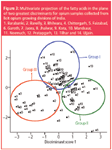
Figure 3: Multivariate projection of the fatty acids in the plane of two greatest discriminants for opium samples collected from licit opium growing divisions of India. 1. Barabanki, 2. Bareilly, 3. Bhilwara, 4. Chittorgarh, 5. Faizabad, 6. Garoth, 7. Jaora, 8. Jhalwar, 9. Kota, 10. Mandsaur, 11. Neemuch, 12. Pratapgarh, 13. Tilhar and 14. Ujjain.
However, some of the samples (Bhilwara, Jhalwar and Kota) of group III were misclassified in group I. Similarly, some of the samples (Tilhar, Chittorgarh) of group II were misclassified in group I and some of the samples (Mandsaur and Neemuch) of group I were misclassified in group II. The misclassification of samples yielded no significant discrimination between the divisions based on the quantitative fatty acid data. For example, opium samples from Garoth and Jhalwar were found in all three groups making it difficult to discriminate one division from the other by means of the fatty acids as a biochemical marker. The same was also observed with other divisions. Based on this discriminant analysis, the results have found to have a 50% predictive value in relation to the source of the opium samples.
A variable reduction process was adopted in order to pinpoint the fatty acid variables that can give a significant discrimination between the licit opium-growing divisions. The mean and standard deviation of the fatty acid variables were calculated between the groups as well as within each group. Variables that provide the largest difference in mean values, but having smallest standard deviation within one group were selected. Variables that have the same or very small difference among the mean values were ignored. The predictive value obtained after this process of variable reduction was found to be less than 50% compared with the predictive value obtained before the variable reduction process. By the above study it is clearly evident that the quantitative fatty acid data is not useful for source identification of Indian opium.
Conclusion
Qualitative analysis of fatty acids such as behenic, stearic and lignoceric acids in Indian opium are valuable biochemical markers for the discrimination of licit opium growing divisions of India. Conversely, quantitative fatty acid data yielded no significant results for the source identification. The methodology developed may find wide application for the determination of fatty acids in opium originating from different licit opium growing countries.
Acknowledgements
The authors are thankful to the Narcotic Commissioner, Ministry of Finance and Government of India for providing permission to collect the opium samples and to the Chief Controller of Factories, New Delhi, India and to the General Managers, GOAW for providing the opium samples. The authors are grateful to the Director, CFSL, Hyderabad for allowing us to perform this work as well as to Dr J. Arunachalam, Head of the National Centre for Compositional Characterization of Materials, Hyderabad for technical help provided in performing the chemometric analysis. Mr M. M. Krishna Reddy is indebted to the Directorate of Forensic Science, New Delhi, India for providing a fellowship at CFSL, Hyderabad and also to CSIR, New Delhi, India for awarding a Senior Research Fellowship to continue this study at the Department of Biochemistry, Osmania University, Hyderabad, India.
M.M. Krishna Reddy is presently working as a scientist in the Analytical Chemistry Section in the Industrial Toxicology Research Centre, Mahatma Gandhi Marg, Lucknow, India.
G. Jayshanker is working as a junior scientific officer in Toxicology at the Central Forensic Science Laboratory (CFSL), Hyderabad, India.
R.B. Sashidhar is with the Department of Biochemistry, Osmania University, Hyderabad, India, as an associate professor. He also served as the Coordinator and Chairman for the Board of Studies of the Forensic Science Unit of the department.
K.M. Varshney is working as Assistant Director with CFSL, Hyderabad, India.
R.K. Sarin is working as Deputy Director with CFSL, Hyderabad, India.
References
1. Statistical Information on Narcotic Drugs, United Nations Office of Drug Control and Crime Preventive, p. 68 (2003).
2. United States of America, National Drug Control Strategy, Reclaiming our Communities from Drugs and Violence, Office of National Drug Control Policy, Executive Office of the President, Washington D.C., p. 16 (1994).
3. Executive Summary on Global Illicit Drug Trends, United Nations Office on Drugs and Crime (UNODC), Vienna, (2003).
4. B. Remberg, A. Nikiforov and G. Buchbauer, Bulletin on Narcotics, XLVI (2), 79–108 (1994).
5. L. Martin et al., UN document # ST/SOA/SER.K/134, UN Secretariat, Vienna (1964).
6. D.S. Lee et al., Analytica Chimica Acta, 358, 163–175 (1998).
7. M.J. Martin et al., Talanta, 54, 291–297 (2001).
8. IUPAC – Standard Methods for the Analysis of Oils, Fats and Derivatives, C. Paquot, A. Hautfenne, Ed. 7th Edition., (Blackwell Scientific, Oxford, UK, 1987).
9. W.R. Morrison and L.M. Smith, J. Lipid Res., 5, 600 (1964).
10. Gardiner W.P. (Ed.), Statistical Analysis, Methods for Chemists — A Software Based Approach. RSC press, Cambridge, UK. p. 313 (1997).
11. B. Remberg et al., Pharmazie, 49, 766– 768 (1994).
12. M.M Krishna Reddy et al., J. Chromatogr. A, 1088, 158–168 (2005).


 with Multi-Angle Light Scattering (MALS) for High-Throughput Protein Refolding")













Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

