Enhancing Mass Spectral Formula Determination by Heuristic Rules
Special Issues
A new approach to enhancing the performance of formula identification of true unknowns beyond high mass and spectral accuracy was evaluated. Three heuristic rules on upper limits and ratios of elements were tested for their effectiveness in filtering out false positive formulas with both high- and low-resolution mass spectrometry data. The rule on elements' upper limits was found to be the most effective one in eliminating incorrect formulas.
Recent advances in the development of new instruments and innovative technology have facilitated the elemental composition determination for unknown identification and compound confirmation significantly. Routinely delivering high resolution to separate ions of interest from background signals and high mass accuracy of 5 ppm or better, many types of new instruments including time-of-flight (TOF), orbital trap, and Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry (MS) systems have been recognized as powerful tools for empirical formula determination. Their capability of formula identification has been further enhanced by utilizing the isotope information (1–4). Of particular importance is the innovative peak-shape calibration that results in mathematically well-defined and symmetrical mass spectral peak shape, which allows exact isotope modeling to improve formula identification performance drastically. Even for an orbital trap system, up to 99% of false positives can be eliminated with the high spectral accuracy achievable, as demonstrated in a recent publication (2). The same technology also made it possible for accurate mass measurements and formula identification with a unit mass resolution quadrupole mass spectrometer (5).
Regardless of this impressive progress in both high mass accuracy and high spectral accuracy, unique formula identification for true unknowns remains a formidable task. This is particularly true, for example, in the identification of natural products and impurities of pharmaceuticals, where there is little knowledge, if any, of the unknowns in terms of possible elements included and their lower and upper limits. To meet the challenge, new approaches known as "seven golden rules to formula identification" were proposed by Kind and Oliver (6,7). Their pioneering work on filtering false positive formulas during elemental composition determination was based upon statistic investigation of the compounds in databases and the first principles of chemistry. Developed from comprehensive analysis of 68,237 unique compounds from both PubChem and Dictionary of Nature Products databases, these seven golden rules can be classified into three categories:
- Chemical elements related ratios (rules 4 and 5), probabilities (rule 6), and their upper limits (rule 1)
- Chemistry principles of Lewis and Senior rules (rule 2) and isotope patterns (rule 3)
- Chemical functional group specific rule for electron ionization MS (rule 7)
While rigorous validations have been done by the statistics on a large formula library and computer simulated spectra, these rules were tested only by limited, experimentally acquired high-resolution data. In this work, the applications of rules 1, 4, and 5 to unknown compound identification using both unit mass resolution and high-resolution data acquired from single-quadrupole, TOF, and orbital trap MS systems will be focused on. Because exact isotope modeling for formula determination (rule 3) has been implemented successfully through software (MassWorks, Cerno Bioscience, Danbury, Connecticut), this investigation will demonstrate which of the three rules will further enhance the formula determination after CLIPS or sCLIPS formula search.
Experimental
Data Acquisition of Unknowns and Calibration Standards: All the data acquisition from both unit mass resolution and high resolution instruments was performed in a profile mode with the threshold set to zero where applicable to ensure accurate isotope measurement. The gas chromatographic (GC)–MS acquisition of tetramethylenedisulfotetramine was performed in a GC–MSD system (Agilent Technologies, Santa Clara, California) in raw scan mode at a scan speed 2^2 (A/D samples = 4) over a mass range of 40–550. After the compound elution at 8.7 min, PFTBA ions were collected for 1 min to be used as calibration standards. In liquid chromatographic (LC)–MS analysis, two separate runs were acquired on an SQD system (Waters Corporation, Milford, Massachusetts). One was for the mixture of atenolol, imipramine hydrochloride, enalapril, and Tyr-Tyr-Tyr, which were calibrated by a set of calibration standards consisting of busprione, reserpine, and erythromycin. The other was for the degradation products of simvastatin (8). Because the m/z value of simvastatin is close enough to the m/z values of its degradation products, simvastatin and its sodium and potassium adducts were conveniently used as calibration standards for the degradants. Additional high resolution LC–MS analyses were performed for probenecid and erythromycin on an Orbitrap MS system (Thermo Fisher Scientific, Madison, Wisconsin) and for Tyr-Tyr-Tyr on an LC–TOF system.
Mass Spectral Calibration: All unit mass resolution data were calibrated by their respective calibration standards. This calibration is the key for unit mass resolution quadrupole instruments to achieve high mass accuracy and exact isotope modeling for CLIPS formula determination. Rigorous description on the calibration was reported previously (9,10). Briefly, the calibration takes both monoisotope m/z values and isotope distribution of calibration standards into consideration. In GC–MS, for example, a set of PFTBA ions, ranged from m/z 68.9 to 501.9, were selected to generate the calibration. Both their m/z values and isotope profiles were calibrated according to the exact mass and theoretically calculated isotope distribution provided by the molecular formula of C9F20N for m/z 501.9. Consequently, their m/z values were corrected and also their peak shape was calibrated to a symmetrical and mathematically well-defined function. This calibration was then applied to unknown data to achieve high mass accuracy and exact isotope modeling for formula determination. For high-resolution data acquired from orbital trap or LC–TOF MS, the calibration was performed on the peak shape only, because high mass accuracy presumably can be obtained from these high-resolution data already.
Formula Determination: Mass tolerance of 15 mDa and 2 mDa were set for the elemental composition determination for unit mass resolution and high resolution data respectively. With no particular restriction, double bond equivalent range was selected from —1 to 50. For molecular ions in GC–MS, an odd electron state was selected, while an even electron state was used for LC–MS data. The upper limits of elements can be set through one of the two ways available in the software. One way is the theoretical maximum, given by the m/z value of the unknown divided by atomic weight for each element. For example, the theoretical possible upper limit of carbon at m/z 240 is 240/12 = 20. The alternative limits are determined by the largest possible chemical elements at various m/z from compound library statistics and proper interpolation between available m/z values (rule 1).
Formula searches for unknowns were performed first based uponly on high mass accuracy and high spectral accuracy and followed by the application of these three rules. These constraints were applied separately one after another so both their individual and cumulative effect on filtering out false positive formulas can be evaluated.
Results and Discussion
Formula identification of the true unknowns requires generous open search conditions so that the upper and lower limits of possible elements should be set to theoretically, the maximum (m/z divided by atomic mass) and zero, respectively. As a result, the number of possible formulas is typically quite large, even though only the most common elements C/H/N/O/S are considered. This problem is further aggravated as the m/z value of unknown compounds increases beyond 500 Da. For example, at 2 ppm mass tolerance, there are 12 possible candidates for buspirone [M+H]+ at m/z 386, but the number of possible formulas increases to 87 for erythromycin [M-H]- at m/z 732. Many of the formula candidates for m/z 386 and 732 can violate fundamental chemistry rules or be statistically improbable.
Formula Reduction by the Three Rules: As demonstrated by the formula search for tetramethylenedisulfotetramine, with five possible elements of C, H, N, O, and S and nine possible elements of C, H, N, O, S, F, P, Cl, and Br (Table I), these false positives can be removed based upon rules 1, 4, and 5. It was found that rule 1 was the most effective one to eliminate wrong formulas, resulting in 42% and 62% reduction from the five and nine elements searches, respectively, while only a 7% and 12% reduction were made accordingly by rules 4 and 5 combined. These results indicate there are more formulas having elements exceeding the maximum number set by rule 1 than those having incorrect element ratios determined by rules 4 and 5. The effectiveness of rule 1 was due to its significantly decreased upper limits for elements N and S in this example. Once rule 1 is applied, the maximum number of N and S was reduced from the theoretically allowable 17 and 7 to 5 and 3, respectively, leading to the elimination of any formula containing more than five N atoms or three S atoms.



Table I
Even though the quantities of false positives filtered out by the rules are an important indication of the overall efficiency for formula reduction, it is more important to examine what spectral accuracy those eliminated formulas have and whether any of the elimination occurs within the top three hits, in which the correct formula usually appears with about 90% probabilities. Indeed, in each of the five-element and nine-element searches shown in Table I, two formulas with spectral accuracy better than 98.5% (Table II) and one formula with spectral accuracy at 98.9% (data not shown) were removed from the top three hits, respectively. Because of their high spectral accuracy and high ranking, these false positives could hardly be distinguished from the correct formula. Their removal by rule 1 from the top three hits leads to more confident compound identification.

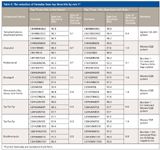
Table II
The Reduction of Formulas on Top Three Hits: With a focus on formula reduction on the top three hits, additional formula search was conducted for a total of seven compounds. They were acquired from either single-quadrupole or high-resolution MS, covering mass range from 240 to 732 Da. Summarized in Table II, out of these eight formula determinations, five had two incorrect formulas filtered out of the top three hits and three had the first hit removed as incorrect formulas. To estimate the impact provided by rule 1 to the differentiation between the first hit and the second hit, the spectral accuracy (SA) difference ΔSA was calculated before and after rule 1 was applied. All ΔSA values from the search with rule 1 show significant increase. Five out of eight ΔSA values increased from 0.3–1.1% to over 1.4%. This difference appears to be too small to be meaningful by conventional wisdom, but it is statistically significant for high-confidence unknown identification due to the exact isotope modeling enabled by peak shape calibration technology. As the best example, the initial search for probenecid resulted in C9H16N7O2S (wrong formula) and C13H20NO4S (correct formula) as the first and second hits, having spectral accuracy of 97.0% and 96.3% respectively. After the search with rule 1 was enabled, both the first hit and third hit from the initial top three were removed and the correct formula C13H20NO4S became the number one hit. As shown clearly in Figure 1, the ΔSA value for this compound increased from 0.7% initially to 4.1%, largely due to the absence of one S atom in the second formula C16H16NO4.


Figure 1
Conclusion
Formula determination for true unknowns can be facilitated by the heuristic rules. The rule on the upper limits of elements (rule 1) was found to be the most effective among the three rules. This rule helps to filter out the majority of false positives. More importantly, it eliminates incorrect formulas from the top three hits obtained by exact isotope modeling. Such reduction of the false positives with high spectral accuracy from the top three hits significantly boosts the confidence of formula determination. With the added capability provided by these heuristic formula rules, the software described here is delivering the most comprehensive and powerful formula identification tool for mass spectrometrists.
Ming Guo is with Cerno Bioscience, Danbury, Connecticut.
References
(1) K. Hobby, R.T. Gallagher, P. Caldwell, and I.D. Wilson, Rapid Commun. Mass Spectrom. 23, 219–227 (2009).
(2) J.C.L. Erve, M. Gu, Y. Wang, W. DeMaio, and R.E. Talaat, J. Am. Soc. Mass Spectrom. 20, 2058–2069 (2009).
(3) K. Hobby,Waters Application notes, Lit Code Number, 720001345EN
(4) C. Koester, (Bruker Daltonik GmbH, Germany). Application: GB, GB, 1999.
(5) M. Gu, Y. Wang, XG. Zhao, and ZM. Gu, Rapid Commun. Mass Spectrom. 20, 764–770 (2006).
(6) T. Kind and O. Fiehn, BMC Bioinformatics 7, 234–243 (2006).
(7) T. Kind and O. Fiehn, BMC Bioinformatics 8, 105–124 (2007).
(8) M. Gu, Cerno Bioscience, Application Notes, Number 105 (2008).
(9) Y. Wang, U.S. Patent No. 6,983,213 (2006).
(10) D. Kuehl and Y. Wang, Spectroscopy "Current Trends in Mass Spectrometry," 38–43, November 2007.













Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Analyzing Vitamin K1 Levels in Vegetables Eaten by Warfarin Patients Using HPLC UV–vis
April 9th 2025Research conducted by the Universitas Padjadjaran (Sumedang, Indonesia) focused on the measurement of vitamin K1 in various vegetables (specifically lettuce, cabbage, napa cabbage, and spinach) that were ingested by patients using warfarin. High performance liquid chromatography (HPLC) equipped with an ultraviolet detector set at 245 nm was used as the analytical technique.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


