Branching Out: Mass Spectrometry and the Shape of Biotherapeutics
Special Issues
Those fond of puns point out that mass spectrometry (MS) has become ever more focused in the last two decades, while at the same time offering ever more information. The dynamic market for biotherapeutics has driven a number of developments, particularly following the paradigm of well-characterized biopharmaceutical products (WCBP) (1,2). Partly as a result of automation and interfacing, those trained in biological or biochemical disciplines now use mass spectrometers routinely. This also means that the sorts of questions asked of MS have changed. Coping with biomolecule heterogeneity is a key challenge, not generally an issue for small molecule drugs. The data complexity means that mass information alone is insufficient. And at the submission stage, regulators are increasingly concerned about tertiary structure and conformation, something that was not previously an analytical requirement (2). Adding polyethylene glycol (PEG) to already heterogeneous molecules to prolong their half-lives in the body raises..
Tandem mass spectrometry (MS-MS) was a revolution in its own right, providing structural information with greater ease. The latest generation of hybrid, tandem mass spectrometer has evolved in an orthogonal dimension by the incorporation of ion mobility. Kanu and colleagues' 2008 prediction (5) that within a few years all mass spectrometers would incorporate an ion mobility cell is being borne out by the acceptance of the technique, and the rapid development of novel uses. The ability to separate biomolecules based purely upon shape in the gaseous phase has already had an impact on the development of biotherapeutics and the understanding of interactions between biomolecules and drug targets (6,7). To examine dynamic tertiary structure, the recent capability of performing ultrafast hydrogen–deuterium exchange (HDX) studies linked to modern MS also has meant that the action of new biotherapeutics candidates can be examined in detail and disambiguated (8,9). H/D exchange and ion mobility were hardly ever associated with routine MS previously, but are now being seen as an essential part of the MS toolkit. Knowing the higher order structure of new biotherapeutics will be a dominant theme and MS is already branching out into this new dimension.
Mass Spectrometry Had to Speed Up
When gas chromatography (GC)–MS was the dominant form of MS, 3-s wide chromatographic peaks were the norm. It was some years before liquid chromatography (LC) reached that capability and subsequently, MS had to evolve once more. Therefore acquisition rates of mass spectrometers have increased steadily, in line with electronics advances. Today, routine assignment of eluted LC–MS peaks can be performed by software based upon accurate mass. For example, oligonucleotide and peptide identities already can be identified automatically by their mass and retention time (for example, with Promass for MassLynx, or BiopharmaLynx software respectively, Waters Corporation, Milford, Massachusetts). Automatic deconvolution of peaks in the mass chromatogram typically is performed in the background using an algorithm-based deconvolution of multiply charged fragment ions to their singly charged value. Software packages then compare the deconvoluted masses of the analytes to the expected masses and the corresponding peaks are putatively assigned. But in the future, this capability will be insufficient and other criteria will be needed for disambiguation.


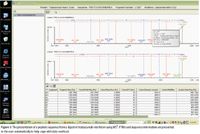
Figure 1
Tools Providing Sequence Information
MS-MS provides detailed sequencing data: precursor ions are fragmented and the data are examined to reconstruct the precursor ion in a manner analogous to re-assembling a puzzle. Fragmentation in mass spectrometers is predictable for a given set of criteria so that software can reconstruct molecules rapidly even though multiply charged species and adduction would make manual interpretation challenging. As data have grown more complex so the techniques and software have had to evolve.


Figure 2
Oligonucleotide failed sequences or peptide sequences can be assigned automatically by using this information. Using automatic peak deconvolution, a 21-mer sequence of RNAi can be confirmed in minutes and the resultant information presented in a graphical format immediately. Truncated failure sequences are typical byproducts of synthesis and can be confidently assigned in this manner. However, in some cases, sequence information is required to disambiguate species. Past methods included selective enzymatic or chemical cleavage (10) or capillary electrophoresis, but LC–MS can provide a more comprehensive technique.


Figure 3
Oligonucleotides that are designed as therapeutic candidates are purified and the remaining contaminants must be characterized. An efficient methodology to comprehensively characterize such biotherapeutics is with ultrahigh-pressure LC–MS (UHPLC–MS) and UHPLC–MS-MS. Importantly, MS-compatible ion-pairing buffers are needed for this type of molecule, requiring dedicated chromatography chemistries. So without the concomitant robustness of the chromatography, the assignment would lack confidence. Having the appropriate chemistries and elution conditions means that certain species are ionized more efficiently and can be detected more easily by MS. The transition of the pharmaceutical industry to drug pipelines dominated by biomolecules has meant that different sets of chromatography chemistries have had to be developed appropriate to the analytical requirements and designed to be compatible with MS.
Oligonucleotide molecules exceeding several thousand daltons in molecular weight might be beyond the range of traps and quadrupole systems. Quadrupole time-of-flight (QTOF)-MS instruments, though, cope with a mass range into the millions of daltons, and have high mass accuracy and resolution, allowing confident assignments. Therefore, higher mass ranges and better mass accuracies are now a defining feature of newer mass spectrometers and will continue to be well into the future.
Coping with Molecular Complexity by Sequence Confirmation
The difficulties in coping with molecular complexity are illustrated by a workhorse technique in biotherapeutics protein characterization: peptide mapping. Regulatory guidelines stipulate that an organization needs to confirm the amino acid sequence and monitor and quantify modifications. Peptide mapping is needed to unambiguously determine posttranslational modifications (PTMs), which can be of very low abundance, can result in a minor mass shift, or can occur in multiple locations. Enzymatic digestion of biotherapeutics (with trypsin, for example) produces predictable peptides that can be examined individually to quantify the PTMs and other changes such as amino acid substitutions.
However, the reduction in average analyte size comes at the price of a corresponding increase in data complexity. Some complexity can be managed by greater chromatographic resolving power available in UHPLC to reduce peptide coelution and analyze them in turn. But with thousands of peptides in a typical 90-min run, many of similar chemical properties, it is impossible to separate them all chromatographically. In the past, mass spectrometers were programmed to use the ion signal intensity to switch to MS-MS mode, known as data-dependent acquisition (DDA). The identity of a molecular ion was then confirmed by matching the fragments to that molecular ion. Despite ever more sophisticated techniques to cope with the switching back and forth, low abundance peptides can still be missed. Even worse, targeted DDA techniques do not provide quantitative information and sometimes the analysis would need to be repeated because of lack of reproducibility. So a robust strategy that provides quantitation and identification simultaneously has productivity advantages for a biotherapeutics developer.
A straightforward technique is to use MSE to generate a comprehensive dataset without preknowledge of the sample. This dataset is achieved by using a QTOF-MS system in MSE acquisition mode to obtain two parallel data sets, one at low collision energy and the other at high collision energy. The resulting raw data file contains parallel data channels with the molecular ions in one, and fragment ions in the other. The data in these channels are linked and the relationships between the molecular ions and the fragments are calculated automatically based upon a number of criteria such as retention time and other cues. So from a single UHPLC injection, MSE provides comprehensive structural information. Because the technique is not biased for signal intensity, the data generated are quantitative. Furthermore, the simplicity of the technique means that ordinary users do not need to be experts in MS to obtain meaningful information. MSE is also a technique that lends itself easily to automation, a boon for the use of mass spectrometers in environments where it is the information which is important, not the details of how MS works.
Having such a comprehensive data set is not the end of the road. To check this information manually would be prohibitively time-consuming and would lead to an unacceptable delay in the development of a biotherapeutic. Therefore, automated software packages to mine MSE data have been invented that automatically display peptide or protein sequences and modifications. For a biotherapeutic, it is important to ensure that a similar product is made with every cell line, or with every batch (11,12). While for small molecule analyses, an extremely high mass accuracy might be sufficient, this is inadequate for a biomolecule that has several PTMs or several glycosylated forms that can affect efficacy. What is important to biotherapeutics manufacturers and regulators is where differences lie, or where anomalies are found in comparison with a control sample.
In a recent study, we were able to show that a product in development did not have the same primary sequence as that intended (19). An examination by LC and optical detection would have been insufficient to show a difference because there would not have been enough discrimination by retention time and UV response. Because an MSE methodology was used with no preconception about the possible variations, the automated mining of the MSE data highlighted the difference in a comparative mode. Had the experiment not been performed with a view toward gaining comprehensive information, the human user would never have known to look for a seemingly insignificant difference in the amino acid sequence. The organization would have risked an expensive and futile scale-up for a substance that was not the one they had intended to make.
The Need for Speed for Dynamic Conformational Analysis
Whereas in peptide mapping, UHPLC has benefited resolution greatly, there are other applications where the raw speed of a UHPLC separation is used to maximum effect. One of these is in HDX, where it is crucial to manage "back-exchange" of deuterium.
The basic principles of HDX are that the amide hydrogens on the backbone of a biomolecule are more exposed in solution and therefore more prone to exchanging with the deuterium in a solution of D2O. Over a time-course experiment, the number of hydrogens that exchange can be measured by MS and therefore, the degree of activity at various sites can be indicated. Certain locations are more prone to exchange because they are less protected. These locations can be identified at the peptide level and then mapped. Thereafter, the relationship to biological activity can be correlated to that three-dimensional map. A leading researcher, Prof. John Engen (Northeastern University, Boston, Massachusetts) has coined the term "HXMS" for this process (13).
A key factor for success is to introduce the peptide mixture as rapidly as possible into a mass spectrometer to manage the back-exchange. Samples are stored at close to 0 °C to preserve the snapshots in time and then injected onto enzyme-digesting columns. The rapidity is important because as soon as the deuterated molecule warms up on the LC column, the back-exchange accelerates. UHPLC robustness is critical when operating at almost 0 °C with nonoptimal chromatography. Studies have shown conclusively that using UHPLC for this type of work considerably aids the identification of crucial sites of activity (14). MS has also needed to evolve to become faster to cope with this type of experiment.
MS has contributed further to this area of tertiary structure by being able to maximize the data available under these pressurized conditions. By using MSE , the HDX study can obtain overlapping fragment ion data to assist in disambiguating where exactly the deuterium uptake has occurred. Biomolecules in an HDX experiment are typically digested with a nonspecific enzyme. The overlap in the peptides can then be used to work out where the exchange took place. The comprehensive nature of the technique means that all of the peptides are measured, without human bias being introduced (8,15).
Because biotherapeutic candidates can have many forms, finding the most efficacious form might need a comprehensive study of the higher order structure (9). The time to market for new biotherapeutics is crucial, therefore, an organization having to screen a number of candidates for efficacy will benefit by using such techniques to reduce development time.
Branches, Stems, and Sugars
Glycosylation is the most common PTM and plays a vital role in the safety and efficacy of recombinant monoclonal antibodies (rmAb). Manufacturers of drug rmAbs must provide regulatory agencies with a detailed assessment of the product glycan microheterogeneity and batch-to-batch consistency. To examine this in detail, glycans can be cleaved enzymatically from the rmAbs and typically are analyzed by LC coupled with a fluorescence detector. With recent developments in UHPLC and sensitivity improvements in QTOF-MS, UHPLC–fluorescence-QTOF-MS can be utilized as a new and improved LC–fluorescence-MS system for rmAb batch-to-batch glycan characterization or profiling at any stage in drug development. The resolution gained by UHPLC makes the glycan assignment based upon the retention times less ambiguous, and also simplifies the fluorescence peak integration. Importantly, new chemistries have had to be developed to be able to efficiently handle this type of sample, for example a UHPLC glycan column packed with sub-2-µm particles for separations by hydrophilic interaction chromatography (HILIC). Drastic improvement in peak resolution and reduction in run times are observed when compared to the conventional HPLC separations. Historically, analytical tools such as matrix-assisted laser desorption ionization (MALDI)-MS and nanoLC–MS were needed for glycan mass profiling and identification in addition to LC–fluorescence but are poor at quantitation. A UHPLC–fluorescence–QTOF system (UPLC/FLR/Xevo Qtof, Waters Corp.) does not have this limitation.
Figure 4 illustrates the UHPLC–fluorescence-QTOF-MS analysis of 2AB-labeled N-linked bovine fetuin glycan. In the fluorescence chromatogram the tri- and tetra-antennary multiply sialylated isomeric glycans (G3S3 and G3S4) are resolved completely at baseline. The increased sensitivity means that with a 10 pmol glycan sample injected the QTOF-MS signals were sufficient for mass profiling and confirmation. Quantitative and qualitative analysis can thus be achieved on a single analytical platform.


Figure 4
Biotherapeutic manufacturers are required to understand variations in batches such as those attributable to changes in rmAb cell culture processing. The methodology applied here illustrates how two different batches of a commercially available humanized recombinant IgG are measurably different. Figure 5 shows the UHPLC–fluorescence-QTOF-MS chromatogram of 2AB labeled glycans released from the IgG, where the most salient difference was the high mannose type (Man 6, 7, 8), present only in batch 2 (Figure 5a). The extracted ion chromatogram (XIC) of these high mannose glycans (Man 5-8) showed that the MS detection was able to verify these glycans by accurate mass measurement (Figure 5b).


Figure 5
Biotherapeutic manufacturers realize that it is critical to develop appropriate analytical tools to characterize glycans to this level of detail. High chromatographic resolution, reproducibility, and MS sensitivity provide a robust and dependable route for an analysis that was incomplete using previous generations of analytical instrumentation.
Glycopeptides and Ion Mobility
Ion mobility MS (IMS-MS) has been applied to biomolecules because the extra dimension of separation means that complexity can be reduced further. In an IMS-MS system, an ion mobility cell replaces a traditional collision cell for an extra dimension of separation. This way, differently shaped ions are separated, even though they might have the same molecular weight. Additionally, IMS can help better detect low-intensity species that would otherwise be masked. The dynamic range improvements for biotherapeutics developers mean that the dramatic increase in heterogeneity and complexity is dealt with in a novel and simple way.
Novel MS systems are always reused in ways that were not previously imagined. An example of this is the use of ion mobility for the rapid characterization of glycosylation in biotherapeutics. Glycosylation of biomolecules is known to play an important part in their activity and being able to rapidly screen for the different forms can provide important information on lot-to-lot heterogeneity (16).
In a time-aligned parallel (TAP) MS-MS experiment (see Figure 6), use is made of the internal architecture of a Synapt HDMS system (Waters Corp.), where the ion mobility cell is flanked by two collision cells. Because each collision cell can be operated independently, different fragmentation conditions can be applied to each one. This means that in the first cell ("Trap"), the glycans can be removed gently with a lower energy. In the mobility cell, all of the glycans and the peptides are separated, and in the second collision cell ("Transfer"), the peptides themselves are fragmented to confirm the peptide sequence. On conventional QTOF mass spectrometers, such an experiment would require separate LC–MS runs, or are performed at a much slower speed. Now the scientist simultaneously obtains information on the location of the glycan as well as confirming that the sequence of the peptide in seconds. Additionally, the methodology applies to both O-linked and N-linked glycans, being equal under a fragmentation regime, despite their different relative abundances.

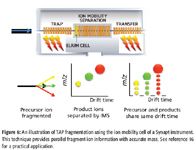
Figure 6
Prototype Work and Advanced Work
In prototype work with a Synapt HDMS system, gas-phase reactions have been performed to simplify the tasks and reduce the steps needed to provide useful information.

Figure 7
One example is recent work on gas-phase HDX, in which, instead of performing the exchange in solution outside of the instrument, it was instead performed inside the mass spectrometer (17). This study demonstrated that it is possible to perform rapid and efficient gas-phase HDX, allowing greater understanding of the dynamic behavior of the proteins. Furthermore, the extent of labeling could be controlled by pressure and speed of the ion-transfer mechanisms. So from this analysis, the detection of conformers present at submillisecond time scales was possible. This differs from techniques where ions are stored (traps) because here all ions are labeled for the same amount of time and the instrumental setup should be readily compatible with online LC. The system architecture lends itself to performing IMS and HDX analysis in tandem so that the same population of ions can be studied in two orthogonal dimensions of conformational detection. This work helps biotherapeutics developers obtain much greater levels of information in less time as compared to HPLC.


Figure 8
Pegylation and Advanced Structural Work
A simple technique for the characterization of PEG and protein conjugates is to use ion mobility to provide a map of the compound mixture. This means that a visual display can be used to characterize a pattern of PEG or biomolecule mixtures quickly, and also can be used to draw only that information that is pertinent while still acquiring everything.
A similar principle of reusing the architecture in novel ways can help with pegylated proteins. The addition of PEG to biomolecule drugs slows down in vivo proteolytic digestion. The alteration of biological activity with PEG also means that this addition can allow the biotherapeutics to be classified as a "different" drug or given orphan status, allowing them to be marketed for a smaller target group of patients (18).
In recent work using gas-phase reaction in the source or in the trap and transfer regions, spectral simplicity was promoted by "charge-stripping" (3,4). The reduction in spectral complexity means that oligomers are resolved and average molecular mass and polydispersities for polymers could be determined. The combination of ion–molecule reaction methodologies with ion mobility separations and TOF-MS provides enhanced sensitivity and specificity for characterizing PEG on the basis of chain lengths and for differentiating low molecular weight impurities. These approaches have enormous potential to characterize PEG materials rapidly and more extensively than before.
Conclusions
New MS capabilities have boosted the type and amount of information available for the development and analysis of biotherapeutics. The initial forays into this area were tentative but appear to have gathered pace, with regulatory interest and biotechnology development driving many analytical areas. The trends have moved very clearly toward an emphasis on conformation and the relationships between biomolecules. However, the data curation has had to keep pace with the increased data overload and therefore sophisticated and automated methodologies with dedicated applications managers have had to evolve to manage the process.
While significant technological strides have been made already (notably in instrumentation to handle conformational analysis and to provide sequence information automatically), we can expect to see technology advance further by providing more detail more rapidly and for postacquisition data software to evolve to meet the need for automated data interpretation.
Joomi Ahn, Vera Ivleva, St John Skilton, and Yingqing Yu are with Waters Corporation, Milford, Massachusetts.
References
(1) FDA/ EMA Guidelines. http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
(2) Guidance for Industry; Comparability Protocols — Chemistry, Manufacturing, and Controls Information. \\CDS029\CDERGUID\5427dft.doc. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070545.pdf
(3) D. Bagal, H. Zhang, and P.D. Schnier, Anal. Chem. 80, 2408–2418 (2008).
(4) A.B. Chakraborty, W. Chen, and J.C. Gebler, Pharm. Tech. 32(7), 80–87 (2008).
(5) A.B. Kanu, P. Dwivedi, M. Tam, L. Matz, and H.H. Hill, Jr., J. Mass Spectrom. 43, 1–22 (2008).
(6) V. Alverdi, H. Mazon, C. Versluis, W. Hemrika, G. Esposito, R. van den Heuvel, A. Scholten, and A.J. R. Heck, J. Mol. Biol. 375, 1380–1393 (2008).
(7) S.Y. Vakhrushev, J. Langridge, I. Campuzano, C. Hughes, and J. Peter-Katalinic, Anal. Chem.
(8) D.Houde, J. Arndt, W. Domeier, S. Berkowitz, and J. R. Engen, Anal. Chem. 81(7), 2644–2651 (2009).
(9) J. Zhang, F.J. Adrián, W. Jahnke, S.W. Cowan-Jacob, A.G. Li, R.E. Iacob, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P.W. Manley, J.R. Engen, G.Q. Daley, M. Warmuth, and N.S. Gray, doi:10.1038/nature08675
(10) Maxam-Gilbert, http://en.wikipedia.org/wiki/DNA_sequencing#Maxam.E2.80.93Gilbert_sequencing)
(11) http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070262.pdf.
(12) http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=2005_register&docid=fr24mr05-5.pdf. Federal Register / Vol. 70, No. 56 / Thursday, March 24, 2005 / Rules and Regulations.
(13) http://www.hxms.com/hxms.htm.
(14) T.E. Wales, K.E. Fadgen, G.C. Gerhardt, and J.R. Engen, Anal. Chem. 80(17), 6815–6820 (2008).
(15) J.L. Mitchell and J.R. Engen, "Protein Analysis with Hydrogen-Deuterium Exchange Mass Spectrometry," in Protein Mass Spectrometry (Elsevier, Amsterdam, 2008).
(16) C.W.N. Damen, W. Chen, A.B. Chakraborty, M. Oosterhout, J.R. Mazzeo, J.C. Gebler, J.H.M. Schellens, H. Rosing, and J.H. Beijnen, J. Am. Soc. Mass Spectrom. 20, 2021–2033 (2009).
(17) K.D. Rand, S.D. Pringle, J.P. Murphy III, K.E. Fadgen, J. Brown, and J.R. Engen, Anal. Chem. 81, 10019–10028 (2009).
(18) Committee for Orphan Medicinal Products Public summary of positive opinion for orphan designation of pegylated recombinant human factor IX for the treatment of haemophilia B. European Medicines Agency; London, 05 June 2009. Doc.Ref.: EMEA/COMP/214529/2009.
(19) H. Xie, A. Chakraborty, J. Ahn, Y. Qing Yu, D.P. Dakshinamoorthy, M. Gilar, W. Chen, S. Skilton, and J.R. Mazzeo, mAbs 2(2) (2010).








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Altering Capillary Gas Chromatography Systems Using Silicon Pneumatic Microvalves
May 5th 2025Many multi-column gas chromatography systems use two-position multi-port switching valves, which can suffer from delays in valve switching. Shimadzu researchers aimed to create a new sampling and switching module for these systems.


Studying Cyclodextrins with UHPLC-MS/MS
May 5th 2025Saba Aslani from the University of Texas at Arlington spoke to LCGC International about a collaborative project with Northwestern University, the University of Hong Kong, and BioTools, Inc., investigating mirror-image cyclodextrins using ultra-high performance liquid chromatography–tandem mass spectrometry (UHPLC–MS/MS) and vibrational circular dichroism (VCD).



