Diving into the Structural Details of In Vitro Transcribed mRNA Using Liquid Chromatography–Mass Spectrometry-Based Oligonucleotide Profiling


The production process of in vitro transcribed messenger RNA (IVT-mRNA)-based vaccines has matured in recent years, partly due to the fight against infectious diseases such as COVID-19. One key to success has been the use of modified, next to canonical, nucleotides and the efficient addition of a Cap-structure and poly A tail to the 5’ and 3’ end, respectively, of this massive biomolecule. These important features affect mRNA stability and impact translation efficiency, consequently boosting the optimization and implementation of liquid chromatography–mass spectrometry (LC–MS)-based oligonucleotide profiling methods for their characterization. This article will provide an overview of these LC–MS methods at a fundamental and application level. It will be shown how LC–MS is implemented in mRNA-based vaccine analysis to determine the capping efficiency and the poly A tail length, and how it allows, via RNA mapping, (i) to determine the mRNA sequence, (ii) to screen the fidelity of the manufactured modifications, and (iii) to identify and quantify unwanted modifications resulting from manufacturing or storage, and sequence variants resulting from mutation or transcription errors.
When dealing with the coronavirus pandemic, academia and pharmaceutical companies have been investing greatly in vaccine development and production during the last two years (1,2). Noticeably, for the first time, vaccines based on messenger RNA (mRNA) have been successfully implemented in disease control. Although this might give the impression that mRNA prophylactics/therapeutics are a recent development,
its foundation was laid down three decades ago when in vitro transcribed mRNA (IVT-mRNA) was shown to be translated following injection into the muscle of mice (3). However, progress in the field of mRNA therapeutics has been slowed owing to the instability of RNA in the omnipresence of RNA-degrading enzymes (so-called ribonucleases or RNases). Promising results showing that IVT-mRNA could be successfully implemented in disease control were first obtained when an antibody response was triggered following intramuscular injection of IVT-mRNA encoding carcinoembryonic antigen into mice (4). Meanwhile, the development of IVT-mRNA drugs/vaccines has seen a gradual progression. For example,
almost 50 IVT-mRNA drugs in cancer immunotherapy entered clinical trials between 2006 and 2020 (5). In addition to its use in infectious diseases and in cancer immunotherapy, IVT-mRNA has gained merits in combatting allergic diseases and in protein-replacement therapies (6,7,8,9,10,11,12).
In drug design, mRNA offers many advantages when compared to the use of DNA or proteins as drugs. IVT-mRNA has only to enter the cytoplasm to enable its processing into a functional protein, avoiding nuclear import restrictions or insertional mutagenesis as in the case for DNA drugs and, hence, tackling a major public concern. Furthermore, the administered IVT-mRNA is only temporarily translated in the cytoplasm, after which it is degraded. In contrast to proteins for which heterologous systems do not necessarily guarantee the correct post-translational modifications (PTMs) or allow a facile production, PTMs are not an issue in the design of IVT-mRNAs. Moreover, IVT-mRNA production is fast, inexpensive, and high-throughput (5), an important feature when a highly infective disease such as COVID-19 emerges. Still, designing IVT-mRNA drugs is not completely devoid of challenges (6,11); as far as naked IVT-mRNA is (i) correctly routed to its target tissue and (ii) not digested prior to reaching the latter tissue, only a minimal amount will be endocytosed. Furthermore, IVT-mRNA might trigger an innate immune response upon complexing with the endosomal toll-like receptors (TLR) 3, 7, and 8 in immune cells, or with the cytosolic receptors called retinoic acid-inducible gene I protein (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) in non-immune cells. Whereas single-stranded RNA (ssRNA) is recognized by TLR7 and TLR8, double-stranded RNA (dsRNA) activates TLR3 and MDA5; RIG-I is stimulated by dsRNA having a 5’-triphosphate. Upon complexation, the released interferon and cytokines will stall mRNA translation, initiate mRNA degradation, and direct antiviral activity (13). These responses might be to a certain extent beneficial in the case of IVT-mRNAs serving as vaccines but they are unintended in a protein-replacement therapy. Despite the stability and immune-response issues, IVT-mRNA design is maturing to the extent that it is increasingly less hindered by these hurdles, and even takes full advantage of these cellular strategies to tune the stability and translation efficiency of IVT-mRNA (14).


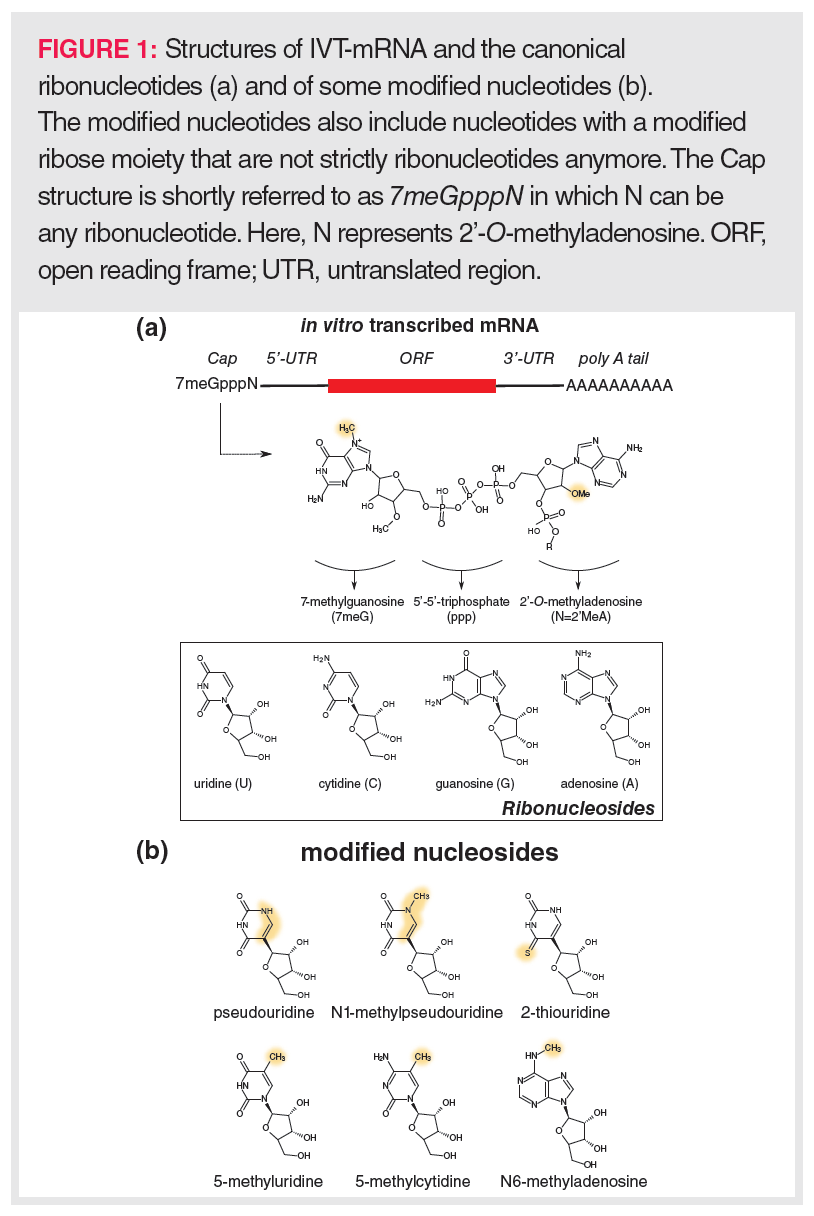
To allow efficient translation, IVT-mRNAs are designed to reflect the structure of natural mRNAs as much as possible (Figure 1[a]). Typically, the coding sequence in an IVT-mRNA (open reading frame; ORF) is flanked by untranslated regions (UTR), and the 3’ and 5’ termini are extended with a 120–150 nucleotide (nt)-long poly A tail and a so-called Cap, respectively. The eukaryotic 5’-Cap structure consists of a 7-methylguanosine (7meG) that is connected to the next nucleotide unit (N) via a 5’-5’-triphosphate linker (7meGpppN). Translation is only possible upon binding of the IVT-mRNA Cap to eukaryotic translation initiation factor 4E (EIF-4E), whereas binding to the decapping enzymes DCP1, DCP2, and DCP3 initiates degradation of the IVT-mRNA. According to the closed-loop model (14), EIF-4E associates with a poly A tail-bound protein complex consisting of EIF-4G and proximal poly-A-binding protein (PABP). Such a closed-loop model enables a fast generation of multiple protein copies—for example, spike glycoprotein in the case of the SARS-COV2 vaccine—via a rolling circle principle. This highlights the importance of the poly A tail for efficient translation next to its role in the stability of mRNA. In addition to the Cap structure and the poly A tail, the UTRs and the coding sequence are important for the IVT-mRNA’s stability, translation efficiency, and the strength of invoking an innate immunity response.
Fine-Tuning IVT-mRNA Synthesis Towards its Pharmaceutical Role
Shorter oligonucleotides (20–50 nt), such as short interfering RNA (siRNA), aptamers, antisense (ASO), and splice-switching oligonucleotides (SSO), are efficiently obtained via the phosphoramidite-based solid-phase synthesis procedure (15). However, this method does not allow the production of full-length mRNAs, which typically have lengths of 1000–4000 nt, or even up to 10,000 nt in the case of self-amplifying mRNA (SAmRNA). Rather, in vitro transcription is necessary to produce mRNAs (6,16,17). Here, the corresponding gene might reside on a plasmid, which necessitates a restriction enzyme-involved linearization of the plasmid. Alternatively, transcription might start from a polymerase chain reaction (PCR) product. In either case, to prevent contamination of the IVT-mRNA by the DNA template, the latter is digested with deoxyribonuclease (DNase) following transcription using, for instance, T7 or SP6 RNA polymerase.
The DNA template encodes the mRNA’s ORF, its UTR regions, and sometimes also the poly A tail (Figure 1[a]). To enhance the IVT-mRNA’s stability and translation efficiency, the 3’-UTR region is often taken from α- or β-globin. For the same purposes, the mRNA’s ORF is generally optimized by replacing rare codons with synonymous frequent codons because relying on more abundantly available transfer RNAs (tRNAs) will augment the translation efficiency. The underlying reason for this is that amino-acylation of tRNAs occurs near the site of translation, which speeds up their recycling.
IVT-mRNAs readily contain modified nucleotides to increase the stability of the IVT-mRNA and/or to circumvent the innate immune system. These modified nucleotides might be based on naturally occurring nucleosides such as pseudouridine, N1-methylpseudouridine, 2-thiouridine, 5-methyluridine, 5-methylcytidine, or N6-methyladenosine (Figure 1[b]), but also include nucleotides that comprise non-natural modifications often targeting the ribose ring. Examples of the latter are nucleotides where 2’-O-methyl substituents are present on the ribose ring.
Post-transcriptionally, the 7meGpppN Cap structure can be formed using recombinant Vaccinia virus-derived capping enzymes, yet the incorporation of the Cap occurs increasingly more co-transcriptionally by adding a synthesized Cap analogue to the mixture of UTP, CTP, GTP, and ATP. Despite capping occurring faster co-transcriptionally than post-transcriptionally, it still yields uncapped mRNAs (generally lower than 10% for commonly used capping procedures) that might trigger the innate immune system. In order to avoid this, free terminal phosphate groups are removed from the IVT-mRNA using a phosphatase.
As mentioned above, the poly A tail can be encoded on the template DNA or it is attached post-transcriptionally via recombinant poly A polymerase. The latter method produces mRNAs with an unfavourable distribution of poly A tail lengths rather than a fixed length, which is optimally between 120 and 150 nt, yet it has the benefit that modified nucleotides can be readily incorporated to enhance protection against poly A-specific nucleases. The efficiency of the Cap incorporation and the poly A tail length determination are of the utmost importance for the quality of the IVT-mRNA, and are typically unravelled via liquid chromatography–mass spectrometry (LC–MS).
The naked IVT-mRNA is not efficiently endocytosed by cells and, therefore, should be encapsulated in a delivery system. Although various delivery systems have been proposed, by far the most efficient to date is based on lipid nanoparticles (LNP). Currently used LNPs contain four components: cationic lipids, phospholipids, cholesterol, and polyethylene glycol (PEG) lipids. Whereas the cationic lipids improve the encapsulation, phospholipids are more readily endocytosed. The assembly of the IVT-mRNA with the LNP is rather challenging and occurs by rapid mixing of all four LNP components in ethanol (18). After adding the pH-4 buffered aqueous solution of IVT-mRNA, the cationic lipids will be charged when contacting the buffer. This will lead to an electrostatic attraction with the negatively charged phosphate backbone of the IVT-mRNA, resulting in the formation of small hydrophobic vesicles. The latter vesicles will fuse to larger-sized vesicles when the cationic lipids are neutralized upon raising the pH of the solution.
The final vesicle size is reached when the PEG-lipid proportion provides a hydrophilic exterior to the vesicle. Important for the quality of the IVT-mRNA/LNP formulation is (i) the vesicle size (< 100 nm), as vesicles may still enlarge after manufacturing, and (ii) the vesicle composition which determines the adjuvant activity of the vesicle, that is, the tendency of the vesicle to induce the innate immune response. The size of the suspended vesicles can be determined via dynamic light scattering (DLS) (19). DLS is based on the scattering of monochromatic, polarized light on suspended vesicles that are subjected to Brownian motion. Due to the latter, the intensity of the scattered light will change over time and is correlated along very short time intervals as the vesicles will not have changed their position too much. However, this so-called autocorrelation quickly decays over time, occurring at a much faster pace for small than for large vesicles as the former vesicles diffuse quicker. Analysis of the lipids constituting the vesicle is readily performed by LC in combination with evaporative light scattering detection (ELSD), charged aerosol detection (CAD), or MS.
Besides the above, an arsenal of analytical methods are involved in the characterization of IVT-mRNA or the IVT-mRNA/LNP formulation (20); however, we will restrict our discussion to LC–MS‑based oligonucleotide profiling methods.
LC–MS Peculiarities in Oligonucleotide Profiling
Current LC methods for poly- and oligonucleotide profiling are mainly based on anion-exchange (AEC), ion pairing reversed phase (IP-RP), or hydrophilic interaction (HILIC) chromatography (21,22,23). As IVT-mRNA characterization relies heavily on MS, this would necessitate the hyphenation of the latter to LC. However, the high molecular weights of IVT-mRNAs (approximately 0.3 × 106–3.5 × 106 Da) prevent MS detection. Therefore, all LC–MS-based IVT-mRNA characterization methods to date involve hydrolyzing the IVT-mRNA towards oligonucleotides or small polynucleotides. Consequently, most LC–MS optimization studies have focused on oligonucleotides, often using higher column temperatures to denature the RNA so that the effects of the secondary structure of RNA on the separation are avoided.
Despite the versatility of LC methods that are employed in oligonucleotide profiling, not all of them are compatible with MS. As elution in AEC employs salt concentration gradients, this method cannot readily be hyphenated to MS, despite the fact that isomeric RNA sequences are often well separated by AEC owing to the polyanionic nature of the RNA phosphate backbone. Nevertheless, electrostatic interactions can be exploited in IP-RP by supplementing an ion-pair reagent (IPR) (for example, an alkylamine) into the mobile phase, while also using a reversed phase stationary phase—the latter often consisting of octadecyl carbon chains (C18). Two mechanisms appear to affect RNA retention in IP-RP: (i) reversed-phase separation of the IPR-RNA complexes and (ii) anion exchange of the RNA molecules onto the IPR that is embedded with its alkyl chain into the reversed phase stationary phase. A counterion, traditionally acetate or bicarbonate in IP-RP, is added to allow buffering of the mobile phase. HILIC is also readily hyphenated to MS. The polyanionic nature of RNA promotes its transfer towards the aqueous stationary phase, whereas the organic solvent-rich mobile phase improves the MS sensitivity. HILIC was first demonstrated for oligonucleotide profiling using a diol-functionalized stationary phase, yet amide groups are currently showing great promise (24,25). Although having the advantage of avoiding the use of IPRs, which yield a memory effect in subsequent LC–MS separations, current HILIC methods still exert poor column stability. This might be partially solved by using higher buffer concentrations (up to 25 mM ammonium acetate or ammonium formate), as higher salt concentrations will aid in establishing a thicker water layer onto the stationary phase support. This improves the solubility of the oligonucleotides into the stationary phase and their retention, yet simultaneously results in a decreased MS signal (26). Although highly promising, HILIC needs further maturation to become a routine LC–MS method for oligonucleotide profiling. Consequently, most oligonucleotide profiling studies are still being performed using IP-RP LC.
The sensitive detection of oligonucleotides via mass spectrometry is a major concern because a wide range of ions is generally formed involving different charge states, isotopes, and alkali adducts. Consequently, an MS peak envelope is created to which often more than 50 channels are contributing (Figure 2). Downscaling the number of charge states needs a good understanding of RNA ion generation in the electrospray ionization (ESI) source of the mass spectrometer. In ESI ionization, an electric field is generated between the metal capillary tip and the mass spectrometer entrance. Analytes in the LC effluent are either charged via the buffer ions present in the mobile phase or via electrochemical reactions at the metal wall of the capillary. As the charges accumulate at the tip of the capillary, a liquid cone is formed in which the charges repel each other. When these electrostatic repulsion forces surpass the surface tension, a Coulomb explosion occurs leading to a spray of droplets, each taking some of the charges towards the mass spectrometer entrance. By heating the ionization source and using a coaxial nitrogen flow, these droplets are continuously shrinking, which results in further Coulomb explosions whenever charges become too concentrated. Finally, the analyte ions will enter the gas phase either by desorption from very small droplets in the case of small molecules (ion evaporation model, IEM), or by inheriting the charges upon complete solvent evaporation of the droplet in the case of compactly folded polymers (charge residue model, CRM). In recent years, a third mechanism, called the chain ejection model (CEM; Figure 3) has been proposed that explains the ionization behaviour of unfolded polymers such as denatured RNA (27). In CEM, one end of the unfolded RNA molecule moves towards the droplet surface where it is charged by the counterion via proton migration while gradually being ejected. Visually, this resembles a tail protruding from a growing tadpole. The latter model explains the wide charge state envelope observed in the MS spectrum of RNA (Figure 2). This suggests that the number of charge states and the average charge state could be lowered by keeping RNA in a tightly folded state, allowing MS ionization via the CRM mechanism. However, a higher average charge state might be necessary when MS/MS fragmentation is envisaged.

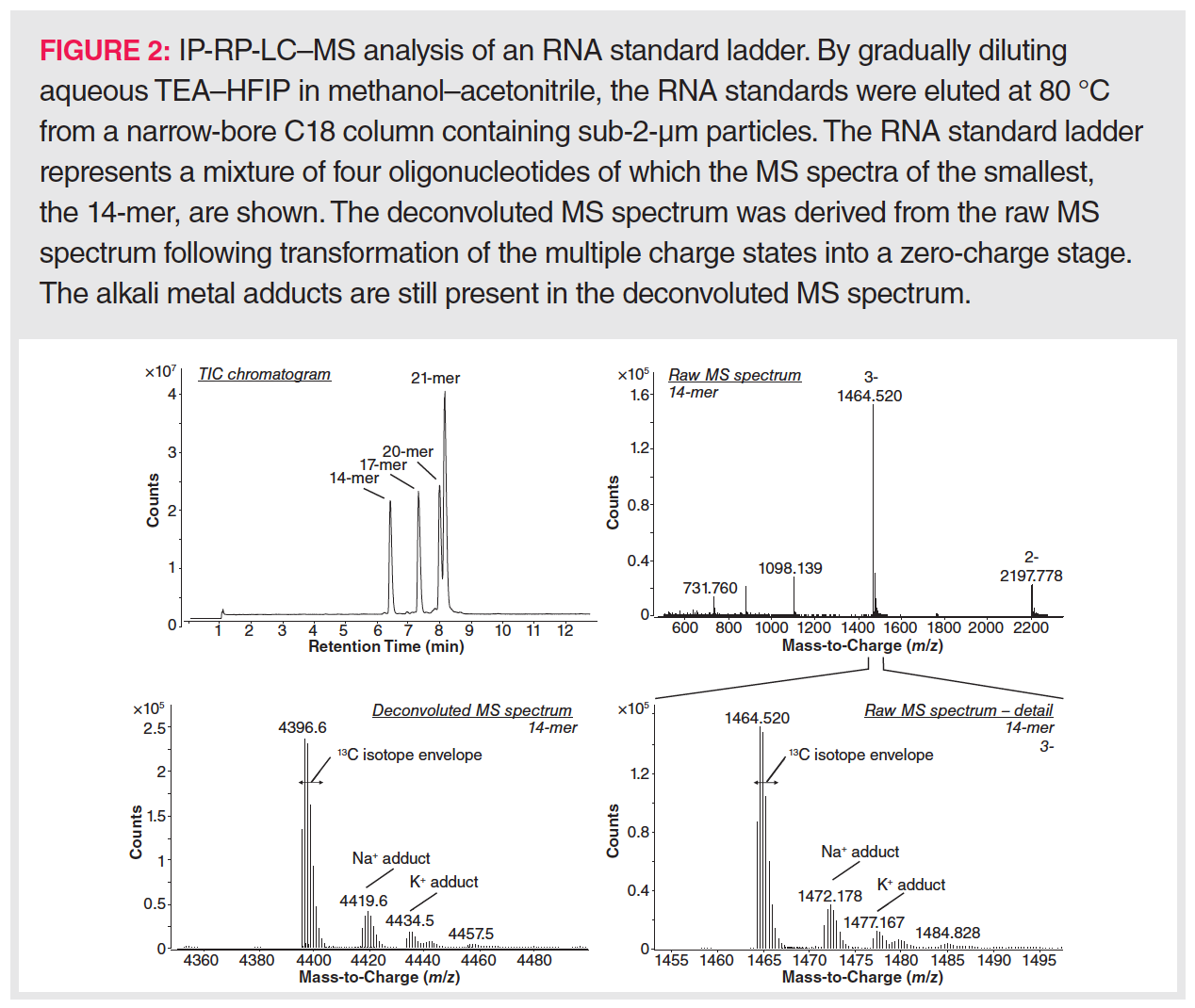
In addition to reduction of the number of charge states, adduct formation should be kept to a minimum to enable a high MS sensitivity. Sources that lead to metal adduction are (i) insufficiently pure solvents and buffers—sometimes leading to a preference for using doubly de-ionized tapwater as mobile phase rather than purchased MS-grade water (28)—(ii) alkali metal salt deposits within the chromatographic system, which can be removed by including a washing step with low-pH mobile phases after each LC run (29), and (iii) any glass surfaces that might be encountered during sample preparation and separation. Concerning the latter issue, glass containers should be avoided, and plastic LC vials and solvent bottles used instead. Furthermore, care should be taken to replace glass solvent filters by plastic filters.
A diminished MS signal and distorted LC separation will result whenever RNA gets degraded or adsorbed onto metal surfaces. The widespread occurrence of RNases is the major source of RNA degradation. Therefore, the use of RNase-free products and an extensive cleaning of all laboratory material are prerequisites for RNA sample preparation. Concerning RNA adsorption to metal surfaces, this is particularly an issue during LC separation. Consequently, the LC instrument should be devoid as much as possible of stainless-steel parts that contact the mobile phase. Such metal surfaces include metal filters, stainless-steel tubing (particularly encountered in ultrahigh-pressure LC [UHPLC]), and the column frits. The latter might comprise up to 50% of all mobile phase-contacting metal surfaces in the LC system. Blocking these metal surfaces (passivation) by so-called sacrificial oligonucleotides improves the reproducibility of MS quantification (30). Alternatively, a bio-inert LC system might be preferred. Meanwhile, bio-inert columns for oligonucleotide profiling are also commercially available, such as polyether ether ketone (PEEK)- or ethylene-bridged siloxane-deactivated stainless steel columns (31). Among the various LC methods for oligonucleotide profiling, metal adsorption might occur less in IP-RP where the many negative charges on the oligonucleotides will likely be blocked by the IPR.
IP-RP as the Prevailing Method for Oligonucleotide Profiling
Alongside the above-described general oligonucleotide profiling issues in LC–MS, the commonly used IP-RP profiling demands paying attention to the IPR and its counterion, as both will affect the separation efficiency as well as the MS sensitivity. The effect of the ion-pairing buffer on the MS sensitivity and the separation efficiency will be discussed separately.
IP-RP Effects on Oligonucleotide Ionization
The use of an ion-pairing buffer promotes ionization via the CEM model (see above), yet the efficiency differs dependent on the type and concentration of IPR and counterion. At least four properties of the ionization process are impacted by the buffer: the CEM process via ion-pair formation, the evaporation efficiency due to crowding of the ESI droplet surface by buffer ions, the electrochemistry of the counterions at the ESI metal capillary wall, and the acid-base reactions between counterion and oligonucleotide in both the condensed and the gas phase. While studies on these mechanisms have made an enormous contribution to our understanding of both the chromatography and MS ionization of IP-RP-based oligonucleotide profiling (32,33,34,35,36,37), it should be stressed that these studies were predominantly based on the use of fluoroalcohols as counterions, which are much weaker acids than the traditionally used carboxylates. Nonetheless, these ionization mechanisms will mostly hold for any weak acid that is used as counterion. The buffer effects on the ionization will be largely independently described. However, it should be kept in mind that the ionization is also affected by interactions among all four buffer-based ionization effects. Furthermore, where appropriate, acid-base reactions between alkylamine and counterion are mentioned in relation to the ionization mechanism.
Effect of Ion-Pair Formation on the CEM Process: The CEM-proposed tail-like ejection of the oligonucleotides is affected by the formation of an ion-pair between the oligonucleotide and the alkylamine within the ESI droplet. As oligonucleotide–alkylamine complexes are more amphiphilic than bare oligonucleotides, they have a higher tendency to move to the droplet surface and being ejected into the gas phase (27,32) (Figure 3). In the dense gas phase just above the droplet surface, oligonucleotide–alkylamine complexes are less vulnerable to ionization (involving a gas phase acid-base reaction) by the counterion than bare oligonucleotides, yielding oligonucleotides that have rather low charge states following dissociation of the complex (34). It must be stressed that complex dissociation occurs while moving away from the droplet towards the less dense gas regions in, for example, the MS pre-analyzer region. In the latter region, the probability of encountering other molecular species is rather limited, preventing any further ionizations via collisions. During complex dissociation, the charges will distribute equally among the oligonucleotide phosphate backbone and the alkylamines, as the proton affinities of the phosphate anion and neutral alkylamine are similar (33).

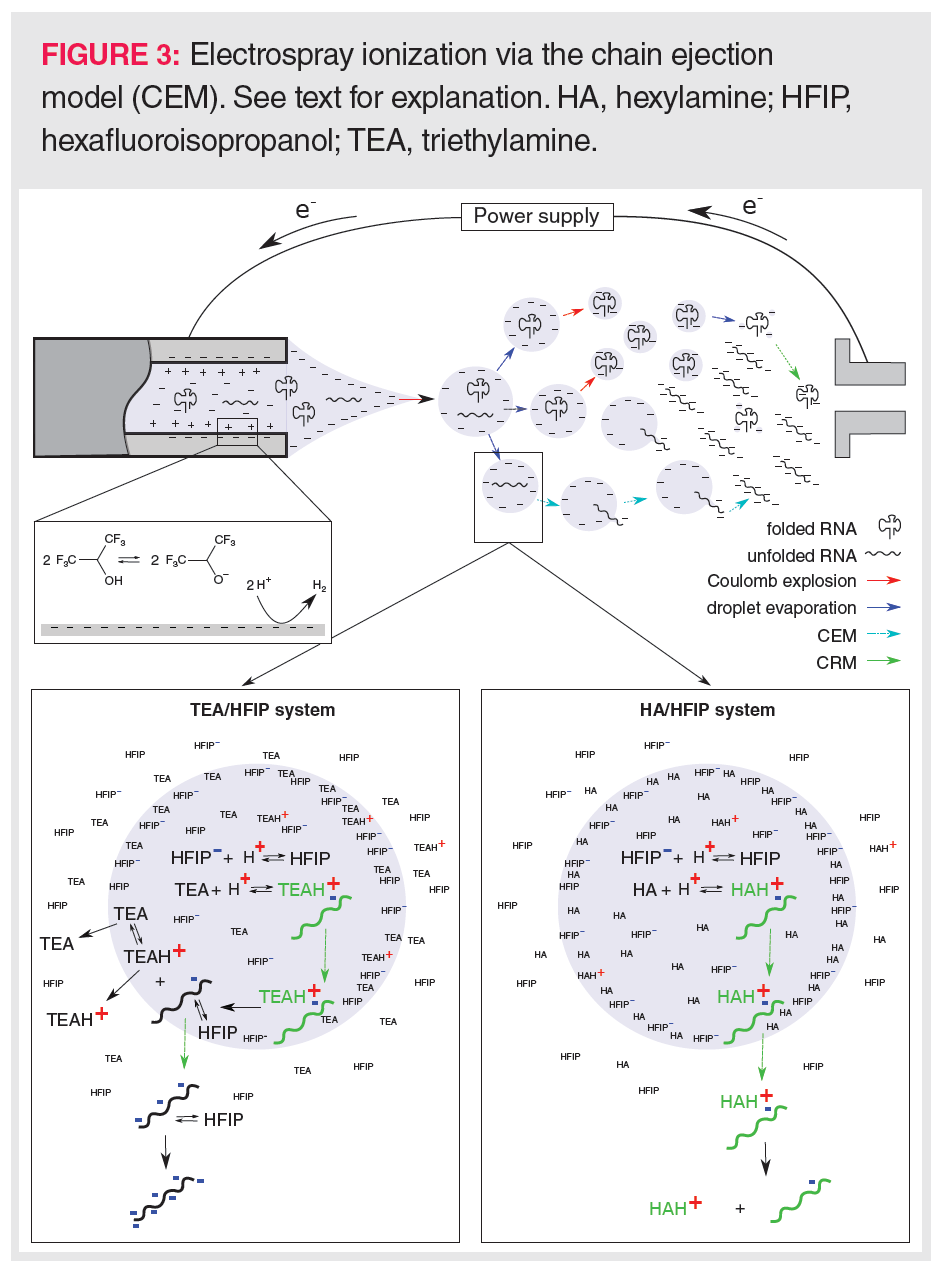
Oligonucleotide–alkylamine complex desorption from the droplet prevails when using linear alkylamines such as hexylamine (HA). HA has a low tendency to evaporate (high Henry’s law partition coefficient), which promotes its adduction to the oligonucleotide (Figure 3). In contrast, the high volatility of triethylamine (TEA) (low Henry’s law partition coefficient) will lead to dissociation of the oligonucleotide–TEA complex just beneath the droplet surface, and the independent evaporation of TEA species and bare oligonucleotides (Figure 3). These bare oligonucleotides are amenable for further deprotonation by the counterion in the gas phase, leading to high charge states. However, an alternative mechanism to explain the low charge state when using HA as IPR was proposed by Basiri et al. (35), who mentioned that linear alkylamines might better keep the oligonucleotide in a folded conformation by neutralizing many of the phosphate backbone charges, and, thus, preventing that charge repulsions would result in more open and elongated forms of the oligonucleotides. Hence, by better conserving the oligonucleotide’s secondary structure, HA would enhance ionization via the CRM model rather than via the CEM model, leading to lower charge states. This mechanism seems plausible taking the Henry’s law partition coefficients of HA and TEA into account. The lower coefficient for TEA implies that TEA more readily evaporates, thereby leaving lower effective TEA concentrations inside the droplet. Subsequently, less TEA will initially enter complex formation than in the case of HA. This would allow highly charged oligonucleotides to unfold more readily when using TEA rather than HA.
Effect of Droplet Surface Crowding by Buffer Ions: Despite the beneficial effect of alkylamines in aiding the transfer and evaporation of oligonucleotides in the ESI droplet, too high an alkylamine concentration might result in ion suppression. Being amphiphilic, the alkylamines mainly reside just beneath the droplet surface (Figure 3). Therefore, when too high alkylamine concentrations are employed, oligonucleotide–alkylamine complexes and oligonucleotides will have to compete with free alkylamines for the droplet surface.
Surface crowding does not only pertain to the alkylamine but also involves the counterion. Being amphiphilic, they also favour the ESI droplet surface. As the counterion is a weak acid, large amounts are often used to buffer the pH of the solution. These high concentrations will crowd the ESI droplet surface and, similarly to high alkylamine levels, will prevent the evaporation of oligonucleotides and oligonucleotides–alkylamine complexes. However, surface crowding does not explain the full behaviour of high counterion levels on the ionization efficiency. A second ionization effect is related to the droplet pH, which is kept low in the presence of high counterion levels. These high proton levels will enhance alkylamine protonation and, thus, the formation of alkylamine–oligonucleotide complexes (36). The latter will support CEM-based ionization affecting the charge state distribution, as described earlier. Noticeably, a low pH might also impact on complex formation by neutralizing many of the oligonucleotide phosphate esters.
Effects Due to Electrochemical Reduction of the Counterion: Next to ion suppression, high counterion concentrations of a weak acid might also lead to ion enhancement. High counterion levels contribute to the ionization of oligonucleotides by providing a large proton pool for reduction to hydrogen at the inner wall of the metal capillary (32) (Figure 3). This electrochemical mechanism removes free protons and therefore enhances the amount of negatively charged counterions in the ESI droplets, increasing the droplet pH and improving oligonucleotide ionization.
Effects Due to Acid-Base Reactions with the Counterion: Finally, the ionization process is also affected by both the solution-based acidity and the gas-phase-based proton affinity of the counterion. Weak acids with higher pKa values offer higher droplet pH values, improving negative ionization of the oligonucleotide. When such acids also have a high proton affinity, they will keep the evaporated oligonucleotides negatively charged in the gas phase. From this perspective, a breakthrough in the hyphenation of IP-RP-LC to MS was the replacement of acetate (pKa 4.8) by hexafluoroisopropanol (HFIP; pKa 9.3) in the commonly used triethylamine (TEA; pKa 11.0) acetate
buffer (37). Compared to an alcohol, the higher acidity of this fluorinated alcohol evolves from the presence of fluorine. The small size and very high electronegativity of fluorine endows the C-F bond with (i) a high dipole moment, increasing the acidity of a nearby acidic function, and (ii) a low polarity resulting in weak dispersion forces explaining the volatility, and the simultaneous lipophobic and hydrophobic properties of fluorinated compounds. As the latter properties suggest, in addition to the stimulating effect of HFIP on oligonucleotide ionization due to its favourable pKa and proton affinity values, HFIP also contributes in other ways to an improved ionization. It improves droplet desorption of the nucleotides by reducing the droplet surface tension and, being a weaker acid and more volatile than acetate, neutral HFIP readily evaporates from the ESI droplet surface. Initially, these beneficial HFIP properties for oligonucleotide ionization were enforced using a high HFIP (400 mM) concentration that was buffered to pH 7 with TEA. Furthermore, methanol rather than acetonitrile was used as organic mobile phase because of the low solubility of HFIP in the latter organic solvent (37). However, despite its high volatility, high HFIP concentrations may still lead to ion suppression by crowding the droplet surface when evaporation is not fast enough (32,34).
IP-RP Effects on Oligonucleotide Retention
Performing the separation at lower pH values boosts ion-pair formation via alkylamine protonation. Therefore, more acidic mobile phases and higher amounts of the alkylamine improve LC retention (38). When compared to a carboxylate, the use of a fluoroalcohol as counterion yields even more retention. This happens because fluoroalcohol-rich mobile phases enhance the sorption of alkylamines into the stationary phase, as the alkylamines cannot dissolve very well in them (36,39). Consequently, apart from improving alkylamine protonation, low pH-buffered IP-RP separations using alkylamine–fluoroalcohol will also stimulate retention by forcing the alkylamine into the stationary phase (due to the necessarily higher fluoroalcohol levels). However, at high alkylamine concentrations, pHs, and/or column temperatures, the alkylamines will form micelles or colloids that will partially absorb the oligonucleotides, leading to their decreased interaction with the stationary phase (22). A ternary mobile phase in which the alkylamine and fluoroalcohol are dissolved in separate solvents might increase the mobile phase stability (40). Recently, it has been shown that concentrations of the alkylamine and the fluoroalcohol that optimize both the retention as well as the ionization of oligonucleotides might be rather low, that is, between 10 and 15 mM and 25 and 50 mM, respectively (33).
It is not only the concentration but also the type of alkylamine and fluoroalcohol that affects retention and ionization of the oligonucleotide (41). The use of alkylamines with a higher number of carbons in their alkyl chain will generally lead to more retention (42,43) and these more hydrophobic alkylamines seem to yield a better LC–MS performance when combined with more hydrophobic fluoroalcohols (35). Many studies have compared the effects of HA and TEA in IP-RP. Both IPR are favoured because they can be more easily cleaned from the instrument than larger IPR such as octylamine or dibutylamine. In addition to yielding oligonucleotides with lower charge states (see above), the use of HA rather than TEA as alkylamine in IP-RP has been repeatedly observed to increase the retention of oligonucleotides (35,39). Therefore, HA might be opted for rather than TEA whenever small oligonucleotides need to be analyzed. Although, IP-RP separations of oligonucleotides are currently heavily reliant on the use of either HA or TEA along with HFIP, alternative alkylamine–fluoroalcohol combinations that further optimize both the separation and ionization efficiencies have been the subject of many recent studies (22,41).
Strategies Towards the RNA Characterization of IVT-mRNA Using LC–MS
Current approaches for the characterization and quantification of the Cap and poly A tail via LC–MS rely on RNases that generate oligonucleotides and, thus, allow isolation of the IVT-mRNA’s 5’- and 3’-ends. Equally so, the confirmation of the mRNA sequence and the detection of modified nucleotides (expected or unexpected) and sequence impurities, such as single nucleotide polymorphisms (SNPs), build upon the combination of RNases and LC–MS profiling. Various RNases with different cleavage preferences are commercially available. Whereas RNases A (cleaves at the 3’ end of cytidine [C] and uridine [U]), T1 (3’ end of guanosine [G]) and U2 (3’ end of any residue) are specific to ssRNA, RNase H recognizes DNA:RNA hybrid duplexes. Two main types of RNase H exist: RNase H1 and RNase H2. RNase H1 needs a DNA:RNA hybrid duplex of at least four adjacent ribonucleotides. RNase H2 will excise any ribonucleotide mismatch in a DNA:DNA duplex—even single ribonucleotide mismatches. Methods comprising RNase digestions are subsequently described below for the analysis of the Cap structure, the poly A tail, and RNA mapping.
Cap Structure Analysis by LC–MS
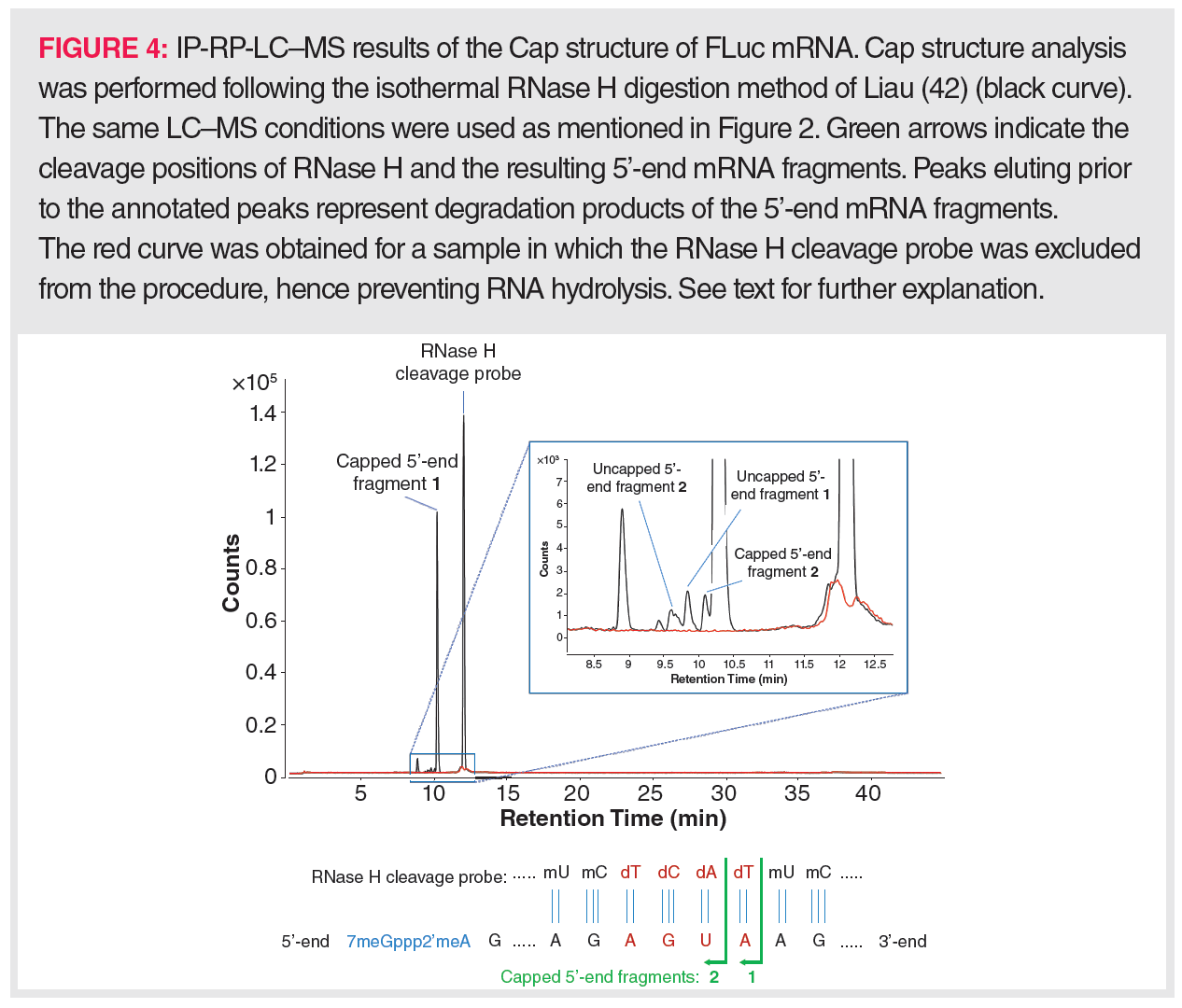
Using ssRNA-specific RNases on IVT-mRNA for Cap isolation yields a mixture of small oligonucleotides, mainly di-, tri-, and tetranucleotides. Upon LC–MS profiling, the capped oligonucleotides can be readily traced based on the specific mass contribution of the Cap structure. Such a method allows different Cap types to be discriminated between, especially when combined with MS/MS fragmentation of the precursor ions (44). However, this method will not allow the uncapped 5’-end-derived oligonucleotides to be distinguished from internal mRNA oligonucleotides with the same sequence and it therefore fails to determine the uncapped proportion in IVT-mRNA. Consequently, attention has been mainly focused on the use of RNase H for Cap analysis. RNase H cleavage necessitates that the IVT-mRNA is first hybridized to a DNA strand, which explains the implementation of a synthesized RNase H cleavage probe in all currently available procedures. This probe should target a sequence that is close to the 5’-end of the IVT-mRNA, so that, following RNase H digestion, a rather short 5’-end oligonucleotide is generated that can be readily separated via LC from the remainder of the IVT-mRNA polynucleotide. Simultaneously, this probe should be sufficiently long, for example, between 15 and 30 nt, to prevent random hybridization. Because digestion of such a long hybrid would lead to a variety of 5’-end-derived oligonucleotides of many different lengths (resulting in a decreased detection sensitivity), the RNase H cleavage probe is typically designed as a gapmer, that is, a chimeric nucleotide sequence that is only partially DNA-based. The most efficient RNase H substrates are RNA:gapmer duplexes in which the DNA sequence occupies a central position in the gapmer. The DNA-flanking gapmer sequences represent modified oligoribonucleotide sequences, for example, 2’-O-methylated oligoribonucleotides, as RNA:RNA duplexes are also vulnerable to RNase H cleavage. If these prerequisites are fulfilled, the RNase H-based Cap isolation procedure can be started and consists of 3 steps: denaturation of the IVT-mRNA at 80–90 °C to remove any secondary structures, hybridization of the RNase H cleavage probe at a temperature below its melting temperature, and RNase H digestion at 37 °C. Assuming that partial unwinding of the mRNA followed by probe hybridization could occur at 50 °C, Liau (45) performed the whole procedure isothermally using thermostable RNase H. The proportion of uncapped versus capped IVT-mRNA could be readily determined by LC–MS as, following digestion, the longer-sized 3’-end of the IVT-mRNA eluted later than its 5’-end. As an example of this isothermal method, the LC–MS profile of the digested mRNA from firefly luciferase (FLuc) is shown in Figure 4; here, a capping efficiency of more than 98% could be determined. Still, dependent on the IVT-mRNA and the designed RNase H cleavage probe, LC conditions might have to be optimized to allow full separation of the IVT-mRNA’s 3’- and 5’-moieties following digestion. Furthermore, the sensitivity of Cap analysis could be improved by purifying the 5’-end from the digest prior to LC–MS analysis. Therefore, Beverly et al. (46) designed a RNase H cleavage probe anchored to a biotin moiety. Via streptavidin-coated magnetic beads, the biotinylated RNase H cleavage probe to which the 5’-end of the IVT-mRNA was hybridized could be isolated from the remainder of the IVT-mRNA digest.

Determining the Poly A Tail Length Distribution by LC–MS
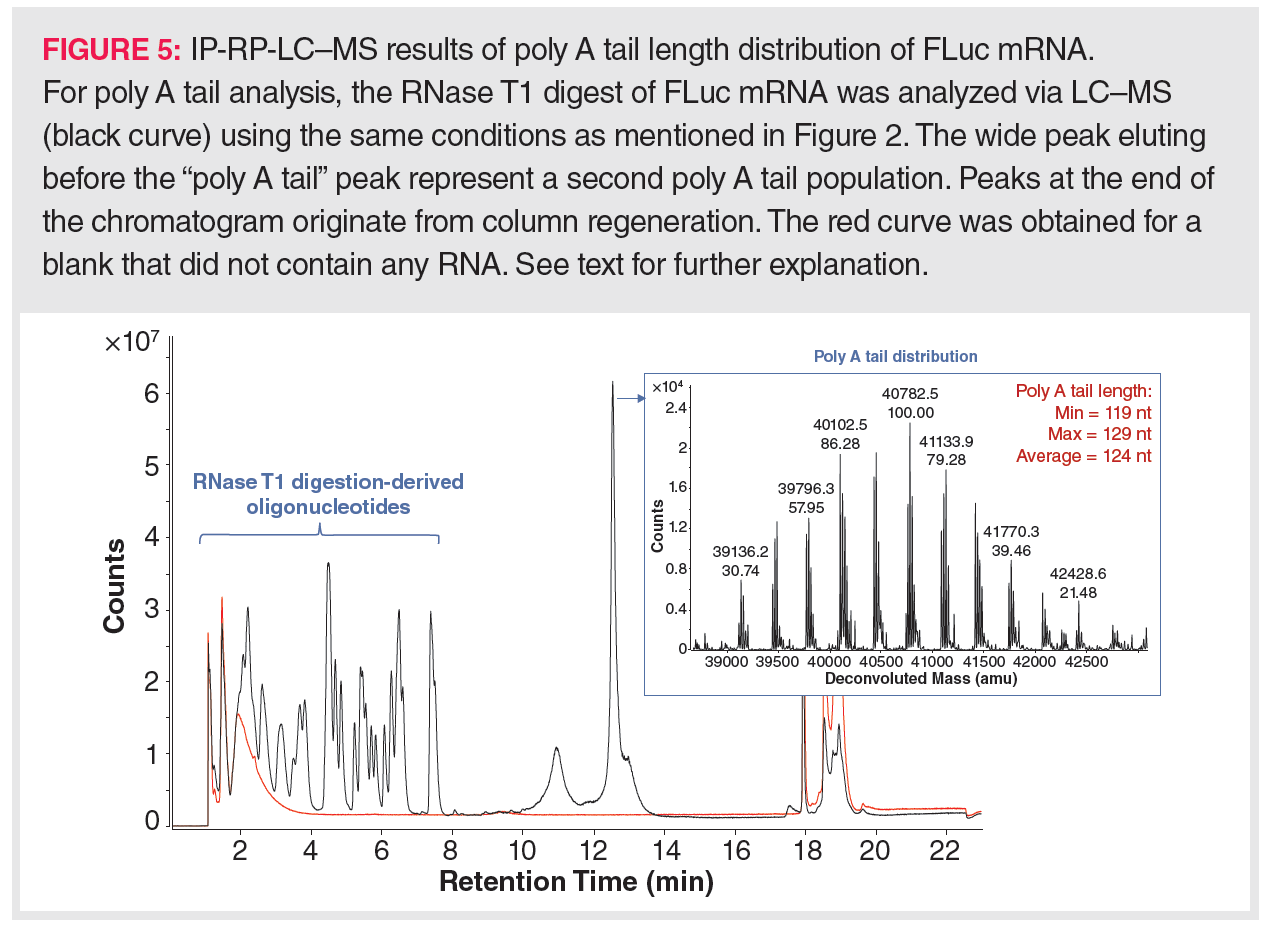
Next to the Cap structure, the poly A tail has received much attention in IVT-mRNA analysis. LC–MS has been implemented in poly A tail analysis due to the inefficiency of current molecular biology methods. For example, deep sequencing cannot handle long repetitive sequences very well, even when the IVT-mRNA has a well-defined poly A tail length, for example, when obtained by encoding the poly A tail in the template DNA. Furthermore, producing the poly A tail post-transcriptionally yields a variety of poly A tail lengths, necessitating an average poly A tail length determination. This can only be handled by LC–MS. Typically, MS detection is preceded by RNA digestion using ssRNA-specific RNases that leave the poly A tail untouched, for example, RNase T1. Such an RNA digestion yields a mixture of single nucleotides and small oligonucleotides, mostly di- up to tetramers, and various poly A species with an average length of 100–150 nt. Hence, the poly A tail species can be readily separated from the oligonucleotides via IP-RP separation. Figure 5 displays the LC–MS results obtained for the poly A tail analysis of the FLuc mRNA as acquired in the author’s laboratory. Here, the different poly A tail species are almost simultaneously eluting, yielding a wide peak. Upon deconvolution of the raw MS spectrum associated with this peak, a frequency distribution of the masses associated with the different poly A tail species can be observed. These masses indicate that the poly A tail length of the analyzed FLuc mRNA ranged between 119 and 129, with an average length of 124. Whenever the elution of the digestion-derived oligonucleotides interferes with the elutions of the poly A tail species, purification of the poly A tail mixture might be desirable prior to LC–MS analysis. In this case, following RNase T1 digestion, the poly A tails can be isolated by hybridization to oligo(dT)25-coated magnetic beads (47,48). Upon denaturation of the oligo(dT)25:poly A tail duplex, the length distribution of the denatured poly A tails can then be analyzed by LC–MS.

Confirming the mRNA Sequence and Detecting Modified Nucleotides by RNA Mapping
More than 170 modified nucleotide structures are currently known to exist; these are comprehensively referred to as the epitranscriptome (49). Some of these modified nucleotides are also used in IVT-mRNA production and therefore need to be considered in determining the mRNA sequence (see Figure 1). However, degradation mechanisms (oxidation and adduction) of IVT-mRNA during manufacturing and storage will yield additional, unintended, modifications to the nucleotides. These modifications will accumulate over time, yielding differently modified IVT-mRNA molecules, ultimately changing the pharmaceutical efficacy of the vaccine/drug. It has been shown, for example, that the oxidation of mRNA (typically, 8-oxo-7,8-dihydroguanosine) inhibits the efficiency of peptide bond formation by more than 1000-fold, regardless of the codon position (50). More recently, LNP lipid-mRNA reactions were described rendering mRNA untranslatable, leading to loss of protein expression. Electrophilic impurities derived from the ionizable cationic lipid component were at the origin of this unfortunate event (51). Tracing such modifications needs a methodology that (i) is sensitive enough to pinpoint modified nucleotides in only a small fraction of the IVT-mRNA pool, (ii) can comprehensively detect all types of modifications, (iii) can handle potentially unknown modifications, and (iv) can quantify the frequencies of the modified nucleotides.
State-of-the-art RNA sequencing via molecular biology techniques involves either nanopore RNA sequencing or they are based on next-generation DNA sequencing (NGS). Nanopore RNA sequencing can allow detection of modified nucleotides, yet it must be trained to associate the electronic signals with the corresponding type of modified nucleotide. Training nanopore RNA sequencing can only be attempted when a large database containing modified RNA sequences is available. However, such databases do currently not exist (52,53).
NGS methods can reveal the sequence of IVT-mRNA following its conversion to complementary DNA (cDNA) via reverse transcription‑PCR (RT-PCR), yet they do not readily provide information on the presence or type of modifications. Such cDNA-based NGS methods are indirect and will only reveal the sequence of the canonical nucleobases in the cDNA. Efforts to detect modified nucleotides via NGS take advantage of the disturbing effect—leading to RT arrest or a nucleotide misincorporation—that many modified nucleotides have on RT-PCR (54). For many modified nucleotides, a disturbing effect on RT-PCR only happens upon a chemical treatment of the RNA with a selective reagent that targets a particular nucleotide or type of modification. Following RT-PCR, sequencing of the aberrant cDNA molecules then allows the position of the modified nucleotide to be pinpointed, yet the identification of the modified nucleotide is often error-prone. In addition, despite being currently popular, these RT-PCR-based methods are cumbersome, tackle a limited set of modified nucleotides, and need validation by LC–MS. Both nanopore RNA sequencing and NGS-based methods handle only a few modifications, do not deal with unknown modifications, and are not able to quantify modifications.
LC–MS approaches that annotate an RNA species based on its sequence or that pinpoint modified nucleotides in the RNA species are based on the profiling of the (oligo)nucleotides following enzymatic digestion or chemical hydrolysis, often complemented by MS/MS fragmentation (15). Complete hydrolysis of RNA will yield a pool of free nucleotides, including all modified nucleotides, which can be subsequently identified and quantified via MS. Although this readily reveals all types of nucleotides and their modifications, this methodology does not provide any information on the position of the modified nucleotides in the mRNA sequence.
RNA digestion with RNases T1, U2, or A has been shown to yield a characteristic oligonucleotide profile that enables RNA characterization (55)—at least in the case of transfer RNA (tRNA) (56) and a CRISPR/Cas9 single guide RNA (57). Optimizing the digestion step is necessary to generate long, overlapping oligonucleotide products. For the latter purpose, it has been shown that including digestions in parallel using different RNases will enhance the generation of overlapping sequences (57). Alternatively, longer sequences upon RNA digestion can be obtained using RNases that are more sequence-specific than RNases T1, U2, or A. Jiang et al. (58) used the endonucleases colicin E5 and mazF, which recognize GU and ACA sequences, respectively. By performing parallel digestions using RNase T1, the authors succeeded in covering 70% of the sequence of a 3000-nucleotide-long mRNA. In addition, the described method allowed low level sequence impurities such as SNPs to be detected and quantified at sub 1% levels. RNA mapping is similar to peptide mapping in that, following digestion, the profiled peptide masses allow an in-depth study of the amino acid sequence and PTMs of the target recombinant protein. These experimental peptide masses can then be matched against in silico generated peptide masses derived from the cloned amino acid sequence. However, in the case of RNA mapping, such a matching procedure might be flawed, as RNA is built from only four nucleotides and so isomeric sequences might be more readily encountered than in the case of proteins. Therefore, in contrast to protein characterization via peptide mapping, the combination of RNA mapping with MS/MS fragmentation is rather essential for RNA characterization.
The gas phase fragmentation behaviour of oligo(ribo)nucleotides is well-known—as well as that of many modified oligonucleotides that are used in designing IVT-mRNAs, such as 2’-O-methyl-derivatized oligoribonucleotides (15). Furthermore, LC–MS conditions can be applied that will yield highly informative MS/MS spectra displaying product ions representing sub-sequences of all possible lengths. This can be achieved, for instance, by fragmenting the highly charged precursor ions, which will augment the range of deprotonated gas phase fragments. To interpret oligonucleotide MS/MS spectra, both freeware and commercial software packages (59–62) have been developed. Using these packages, sequences might be obtained for oligonucleotides up to a length of approximately 25 nucleotides. RNases will yield oligonucleotides that are often much shorter than 25 nucleotides, thereby making RNase-derived oligonucleotides amenable to MS/MS sequencing. However, the question arises as to what extent sequences obtained for these small‑length oligonucleotides are sufficient to reassemble the full sequence of a complete IVT-mRNA. At least in the case of small RNA oligonucleotides (length up to 100 nt), the full sequence and its modifications can be obtained using parallel digestions as described above (57).
An ingenious approach towards LC–MS-based de novo sequencing of RNA starts with generating a ladder of the RNA sequence via partial digestion (63). This method implies (i) that cleavage occurs randomly throughout the full RNA sequence and (ii) the presence of either the 5’- or 3’-end in the cleaved product. Following mass-based ordering of all 5’- or 3’-end-comprising partial sequences, the full RNA sequence can then be derived by considering the mass differences between subsequent partial sequences in the ladder. To easily annotate partial sequences still having a 5’- or 3’-end, a hydrophobic linker can be attached that will lead to a considerable retention shift so that the derivatized sequences elute separately from other oligonucleotides representing internal sequences of the RNA. Up to now, this method has been applied to 20–22-nt-long oligonucleotides, hence, targeting oligonucleotides of lengths similar to those in RNA mapping. Despite LC–MS-based sequencing of RNA having progressed a lot during the last decade, further maturing is necessary to handle polymer lengths of a few thousand nucleotides that are typical for IVT-mRNA.
Conclusions and Perspectives
The COVID-19 pandemic has boosted both the development of mRNA-vaccines and their official approval for prophylactic use by health authorities worldwide. Despite initial distrust, the effectiveness of these vaccines has been shown to downsize the number and severity of infections, therefore changing the adverse opinion of many. However, IVT-mRNA drug development and application are in their infancy, particularly for analytical scientists that need to develop methods to analyze IVT-mRNA. From a structural perspective, IVT-mRNA is to a certain extent still a black box and is not as well characterized as biologics such as monoclonal antibodies. Furthermore, there is a lack of knowledge as to what criteria will be most decisive to the efficacy and safety of IVT-mRNAs. The principal analytical methods that are of use to date are quickly evolving. For example, LC–MS methods for oligonucleotide profiling are currently mainly based on IP-RP. Consequently, much effort is being focused on understanding the effect of the IPR and counterion on separation and ionization efficiency to further optimize IP-RP for oligonucleotide profiling by, for example, the implementation of new IPR/counterion combinations. Simultaneously, part of the RNA-based LC–MS community are turning away from IP-RP because of the memory effects of the IPR in MS, and are instead further developing HILIC. The resolution of one‑dimensional (1D) separations might prove insufficient, especially in the case of RNase digests. Therefore, a renewed interest in multidimensional LC(–MS) will emerge. In that respect, early work on comprehensive two-dimensional LC (2D-LC) for oligonucleotide profiling incorporating HILIC in the first dimension and IP-RP in the second dimension has been described already a decade ago (64). More recently, 2D and three-dimensional (3D)-LC setups incorporating online RNase digestion have appeared in the scientific literature (65). Stay tuned as much more is to come.
LC–MS method development will also be steered by changes in IVT-mRNA manufacturing. For example, rather than attaching the poly A tail using poly A adenylase, the poly A tail could be encoded in the gene, resulting in a poly A tail with a fixed length, thereby alleviating the sensitivity issues that accompany the analysis of a large variety of poly A tails. At the same time, sensitivity issues might eventually become more important in determining the capping amount as capping approaches become increasingly more efficient. Nonetheless, robust Cap structure and poly A tail analysis methods exist that completely contrast the field of RNA mapping—the latter being still in its infancy—compared to peptide mapping. Here, the sequencing of a complete IVT-mRNA and the in-depth study of product-related variants and impurities has not yet been published. Many creative approaches are being generated that are currently only implementable for the RNA mapping of rather small oligo or polynucleotides. More analytical challenges are ahead of us when self-amplifying and circular mRNA mature, as these will be formulated at low concentrations demanding for analytics at ultimate sensitivity. Clearly, analytical scientists must accelerate the invention of new analytical technologies to come up to speed with the field. As we are at the forefront of a new era in drug development, exciting times are ahead of those who work in analytical science.
References
- R. Verbeke, I. Lentacker, S.C. De Smedt, and H. Dewitte, J. Control Release 333, 511–520 (2021).
- L. Schoenmaker, D. Witzigmann, J.A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot, and D.J.A. Crommelin, Int. J. Pharmaceutics 601, 120586 (2021).
- J.A. Wolff, R.W. Malone, P. Williams, W. Chong, G. Ascadi, A. Jani, and P.L. Feigner, Science 247, 1465–1468 (1990).
- R.M. Conry, A.F. LoBuglio, M. Wright, L. Sumerel, M.J. Pike, F. Johanning, R. Benjamin, D. Lu, and D.T. Curiel, Cancer Res. 55, 1397–1400 (1995).
- J.D. Beck, D. Reidenbach, N. Salomon, U. Sahin, Ö. Türeci, M. Vormehr, and L.M. Kranz, Molecular Cancer 20, 69 (2021).
- U. Sahin, K. Karikó, and Ö. Türeci, Nature Rev. Drug Discovery 13, 759–780 (2014).
- T. Schlake, M. Thran, K. Fiedler, R. Heidenreich, B. Petsch, and M. Fotin-Mleczek, Molecular Therapy 27, 773–784 (2019).
- L. Van Hoecke and K. Roose, J. Translational Medecine 17, 54 (2019).
- R. Verbeke, I. Lentacker, S.C. De Smedt, and H. Dewitte, Nano Today 28, 100766 (2019).
- J.A. Kulkarni, D. Witzigmann, S.B. Thomson, S. Chen, B.R. Leavitt, P.R. Cullis, and R. van der Meel, Nature Nanotechnology 16, 630–643 (2021).
- B. Baptista, R. Carapito, N. Laroui, C. Pichon, and F. Sousa, Pharmaceutics 13, 2090 (2021).
- N. Chaudhary, D. Weissman, and K.A. Whitehead, Nat. Rev. Drug Discov. 20, 817–838 (2021).
- N. Pardi, M.J. Hogan, F.W. Porter, and D. Weissman, Nat. Rev. Drug Discov. 17, 261–279 (2018).
- S. Linares-Fernández, C. Lacroix, J.-Y. Exposito, and B. Verrier, Trends Mol. Med. 26, 311–323 (2020).
- S. Pourshahian, Mass Spectrom. Rev. 40, 75–109 (2019).
- S. Oh and J.A. Kessler, Methods 133, 29–43 (2018).
- J. Whitley, C. Zwolinski, C. Denis, M. Maughan, L. Hayles, D. Clarke, M. Snare, H. Liao, S. Chiou, T. Marmura, H. Zoeller, B. Hudson, J. Peart, M. Johnson, A. Karlsson, Y. Wang, C. Nagle, C. Harris, D. Tonkin, S. Fraser, L. Capiz, C.L. Zeno, Y. Meli, D. Martik, D.A. Ozaki, A. Caparoni, J.E. Dickens, D. Wessman, K.O. Saunders, B.F. Haynes, G.D. Sempowski, T.N. Denny, and M.R. Johnson, Translational Res. 242, 38–55 (2021).
- M.D. Buschmann, M.J. Carrasco, S. Alishetty, M. Paige, M.G. Alameh, and D. Weissman, Vaccines 9, 65 (2021).
- T. Terada, J.A. Kulkarni, A. Huynh, S. Chen, R. van der Meer, Y.Y.C. Tam, and P.R. Cullis, Langmuir 37, 1120–1128 (2021).
- C.A. Challener, BioPharm International 35, 10–15 (2022).
- S. Studzińska, Talanta 176, 329–343 (2018).
- J.M. Sutton, G.J. Guimaraes, V. Annavarapu, W.D. van Dongen, and M.G. Bartlett, J. Am. Soc. Mass Spectrom. 31, 1775–1782 (2020).
- A. Goyon, P. Yehl, and K. Zhang, J. Pharmaceut. Biomed. Anal. 182, 113105 (2020).
- R.N. Easter, K.K. Kreoning, J.A. Caruso, and P.A. Limbach, Analyst 135, 2560–2565 (2010).
- A. Demelenne, M.-J. Gou, G. Nys, C. Parulski, J. Crommen, A.-C. Servais, and M. Fillet, J. Chromatogr. A 1614, 460716 (2020).
- M. Huang, X. Xu, H. Qiu, and N. Li, J. Chromatogr. A 1648, 462184 (2021).
- H. Metwally, Q. Duez, and L. Konermann, Anal. Chem. 90, 10069–10077 (2018).
- B. Wei, J. Wang, L. Cadang, A. Goyon, B. Chen, F. Yang, and K. Zhang, J. Chromatogr. A 1665, 462839 (2022).
- R.E. Birdsall, M. Gilar, H. Shion, Y.Q. Yu, and W. Chen, Rapid Commun. Mass Spectrom. 30, 1667–1679 (2016).
- G.J. Guimaraes, J.M. Sutton, M. Gilar, M. Donegan, and M.G. Bartlett, J. Pharmaceut. Biomed. Anal. 208, 114439 (2021).
- S. Fekete, LCGC Europe 34, 245–248 (2021).
- Z. Wu, W. Gao, M.A. Phelps, D. Wu, D.D. Miller, and J.T. Dalton, Anal. Chem. 76, 839–847 (2004).
- G.J. Guimaraes and M.G. Bartlett, Future Sci. OA FSO753 (2021).
- B. Chen, S.F. Mason, and M.G. Bartlett, J. Am. Soc. Mass Spectrom. 24, 257–264 (2013).
- B. Basiri, H. van Hattum, W.D. van Dongen, M.M. Murph, and M.G. Bartlett, J. Am. Soc. Mass Spectrom. 28, 190–199 (2016).
- L. Gong and J.S.O. McCullagh, Rapid Commun. Mass Spectrom. 28, 339–350 (2014).
- A. Apffel, J.A. Chakel, S. Fischer, K. Lichtenwalter, and W.S. Hancock, Anal. Chem. 69, 1320–1325 (1997).
- M. Gilar, K.J. Fountain, Y. Budman, J.L. Holyoke, H. Davoudi, and J.C. Gebler, Oligonucleotides 13, 229–243 (2004).
- G. Vanhoenacker, C. Lecluyse, G. Debyser, P. Sandra, and K. Sandra, Agilent Application Note Biopharma 5994-2957EN (2021).
- N. Li, N.M. El Zahar, J.G. Saad, E.R.E. van der Hage, and M.G. Bartlett, J. Chromatogr. A 1580, 110–119 (2018).
- A. Kaczmarkiewicz, Ł. Nuckowski, S. Studzińska, and B. Buszewski, Crit. Rev. Anal. Chem. 49, 256–270 (2019).
- V.K. Sharma, J. Glick, and P. Vouros, J. Chromatogr. A 1245, 65–74 (2012).
- S. Studzińska, R. Rola, and B. Buszewski, J. Pharmaceut. Biomed. Anal. 138, 146–152 (2017).
- N. Muthmann, P. Špaček, D. Reichert, M. van Dülmen, and A. Rentmeister, Methods in press (2022).
- B. Liau, Agilent Application Note 5994-3984EN (2021).
- M. Beverly, A. Dell, P. Parmar, and L. Houghton, Anal. Bioanal. Chem. 408, 5021–5030 (2016).
- M. Beverly, C. Hagen, and O. Slack, Anal. Bioanal. Chem. 410, 1667–1677 (2018).
- B. Liau, Agilent Application Note 5994-3005EN (2022).
- Y. Saletore, K. Meyer, J. Korlach, I.D. Vilfan, S. Jaffrey, and C.E. Mason, Genome Biol. 13, 175 (2012).
- S.H. Boo and Y.K. Kim, Experimental & Molecular Medicine 52, 400–408 (2020).
- M. Packer, D. Gyawali, R. Yerabolu, J. Schariter, and P. White, Nat. Commun. 12, 6777 (2021).
- D.R. Garalde, E.A. Snell, D. Jachimowicz, B. Sipos, J.H. Lloyd, M. Bruce, N. Pantic, T. Admassu, P. James, A. Warland, M. Jordan, J. Ciccone, S. Serra, J. Keenan, S. Martin, L. McNeill, E.J. Wallace, L. Jayasinghe, C. Wright, J. Blasco, S. Young, D. Brocklebank, S. Juul, J. Clarke, A.J. Heron, and D.J. Turner, Nat. Methods 15, 201–206 (2018).
- W. Stephenson, R. Razaghi, S. Busan, K.M. Weeks, W. Timp, and P. Smibert Cell Genom. 2, 100097 (2022).
- M. Helm and Y. Motorin, Nat. Rev. Genetics 18, 275–291 (2017).
- B. Solivio, N. Yu, B. Addepalli, and P.A. Limbach, Anal. Chim. Acta 1036, 73–79 (2022).
- M. Hossain and P.A. Limbach, RNA 13, 295–303 (2007).
- A. Goyon, B. Scott, K. Kurita, C.M. Crittenden, D. Shaw, A. Lin, P. Yehl, and K. Zhang, Anal. Chem. 93, 14792–14801 (2021).
- T. Jiang, N. Yu, J. Kim, J.-R. Murgo, M. Kissai, K. Ravichandran, E.J. Miracco, V. Presnyak, and S. Hua, Anal. Chem. 91, 8500–8506 (2019).
- H. Nakayama, M. Akiyama, M. Taoka, Y. Yamauchi, Y. Nobe, H. Ishikawa, N. Takahashi, and T. Isobe, Nucleic Acids Res. 37, e47 (2009).
- P.J. Sample, K.W. Gaston, J.D. Alfonzo, and P.A. Limbach, Nucleic Acids Res. 43, e64 (2015).
- S. Wein, B. Andrews, T. Sachsenberg, H. Santos-Rosa, O. Kohlbacher, T. Kouzarides, B.A. Garcia, and H. Weisser, Nat. Commun. 11, 926 (2020).
- L. D’Ascenzo, A.M. Popova, S. Abernathy, K. Sheng, P.A. Limbach, and J.R. Williamson, Nat. Commun. 13, 2424 (2022).
- N. Zhang, S. Shi, T.Z. Jia, A. Ziegler, B. Yoo, X. Yuan, W. Li, and S. Zhang, Nucleic Acids Res. 47, e125 (2019).
- Q. Li, F. Lynen, J. Wang, H. Li, G. Xu, and P. Sandra, J. Chromatogr. A 1255, 237–243 (2012).
- A. Goyon, B. Scott, K. Kurita, C. Maschinot, K. Meyer, P. Yehl, and K. Zhang, Anal. Chem. 94, 1169–1177 (2022).
About The Authors
Kris Morreel is Senior Scientist LC–MS at RIC group (Kortrijk, Belgium) and Visiting Professor at Ghent University (Ghent, Belgium).
Ruben t’Kindt is Head of LC–MS at RIC-group (Kortrijk, Belgium).
Griet Debyser is Scientist LC–MS at RIC group (Kortrijk, Belgium)
Stefanie Jonckheere is Senior Research Associate LC–MS at RIC group (Kortrijk, Belgium)
Pat Sandra is Founder and Advisor of the RIC group (Kortrijk, Belgium) and Emeritus Professor of Ghent University (Ghent, Belgium).
About The Column Editor
Koen Sandra is the editor of “Biopharmaceutical Perspectives”. He is CEO at RIC group (Kortrijk, Belgium) and Visiting Professor at Ghent University (Ghent, Belgium). He is also a member of LCGC Europe’s editorial advisory board.
Direct correspondence about this article to amatheson@mjhlifesciences.com



 Columns")


")








Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

