Dispersive Miniaturized Solid-Phase Extraction Using the CIM-81 Metal-Organic Framework and Gas Chromatography–Mass Spectrometry to Determine Personal Care Products in Waters
Special Issues
In this study, the analysis of contaminants found in environmental waters and originating from personal care products is addressed using metalorganic frameworks (MOFs) in combination with liquid chromatography (LC). This work expands the use of MOFs from gas chromatography to LC and also meets the requirements of green analytical chemistry.
The use of metal-organic frameworks (MOFs) in miniaturized extraction approaches is already successful in analytical sample preparation, particularly when combined with liquid chromatography. This study shows the determination of several semivolatile personal care products with the CIM-81 MOF, but in combination with gas chromatography and mass spectrometry detection, to expand the applications while still meeting the requirements of green analytical chemistry.
Personal care products (PCPs) are organic compounds belonging to the group of emerging contaminants, which have shown toxic effects in humans and other living organisms (1,2). There are six groups of PCPs: ultraviolet (UV) filters, preservatives, disinfectants, musk, insect repellents, and siloxanes, with the classification depending on their use, except for siloxanes, which are classified based on their structure. PCPs are present in a wide number of everyday products, such as gels, lotions, and cosmetics. Given their extensive use, it is clear that high amounts of PCPs are released continuously into the environment, mainly into river, lake, and seawaters, where they are diluted. Accurate monitoring of PCPs in environmental samples is undoubtedly important, requiring efficient methodologies of preconcentration and detection.
The determination of nonvolatile PCPs is normally accomplished using high performance liquid chromatography (HPLC) (3–7), whereas gas chromatography (GC) is utilized for volatile or semivolatile PCPs (8–11). Among microextraction and preconcentration techniques, it is important to highlight the increased utilization of miniaturized solid-phase extraction (µSPE) methods because of their advantages: simplicity, low cost, low consumption of solvents, low time requirements, and low amounts of sorbent needed (<500 mg). The dispersive mode (D-µSPE) is probably the simplest approach, with the sorbent added directly to the aqueous sample under gentle stirring, and it is usually accompanied by high enrichment factors (12). D-µSPE, like many other analytical extraction methods, has benefited from recent incorporation of novel materials as sorbents. Among materials tested successfully, molecularly-imprinted polymers (13), graphene (14), multi-walled carbon nanotubes (15), metallic nanoparticles (16), and metal-organic frameworks (17) can be highlighted.
Metal-organic frameworks (MOFs) are crystalline polymers with a three-dimensional structure (18). MOFs result from the autoassembly of metal ions (or metal clusters) and organic linkers through coordination bonds. These materials are characterized by their high porosity and tuneability, quite low crystal densities, and adequate thermal and mechanical stabilities, combined with high surface area (19). These properties fulfil many of the requirements for sorbents in efficient extractions, and thus MOFs have been successfully incorporated into a number of microextraction strategies (20,21), with the majority of applications linked to D-µSPE (12). Thus, different bare MOFs, such as HKUST-1(Cu) (22), MIL-101(Fe) (23), MIL-101(Cr) (24), MIL-53(Al) (25), TMU-5 (26), and UiO-66 (27), among others, have been used as sorbents in D-µSPE for the extraction of different pollutants.
Most applications of bare-MOFs or MOF-based composites as sorbents in D-µSPE are carried out in combination with chromatographic techniques, and among them, around 80% of the publications use high performance liquid chromatography (HPLC) or ultrahigh-pressure LC (UHPLC) (28,29). The reported applications normally focus on pollutants (such as polycyclic aromatic hydrocarbons, pyrethroids, polychlorinated diphenyl ethers, and hormones) in environmental samples (30–33). In contrast, applications in combination with GC are scarce (34,35). In fact, to date there has not been any study using MOFs as sorbents in D-µSPE-GC for PCPs.
The [Zn2(tz)2(bdc)] MOF, termed CIM-81 in a previous article of our research group (36), is a pillared-layer network constructed from a terephthalic carboxylate ligand (H2bdc) bridging the 2D layers formed by the Zn(II) metal ions and the 1,2,4-triazole (Htz) motifs. The main advantage of using MOFs with mixed ligands (like CIM-81) in D-µSPE, versus MOFs presenting a single type of ligand, is the increasing number of analyte–MOF interactions that can occur. The extraction efficiency of CIM-81 in D-µSPE for several endocrine disrupting chemicals followed by UHPLC with ultraviolet detection (UHPLC-UV) was demonstrated in a previous study (36). Thus, the main objective of the present study was to develop an analytical application for semivolatile PCPs utilizing this successful CIM-81 MOF in D-µSPE, and in combination with GC combined with mass spectrometry (GC–MS). As a parallel goal, this study aims to expand the analytical applications of MOFs for volatile or semivolatile analytes, while encouraging an increased use of GC when utilizing this microextraction method (D-µSPE) based on MOFs.
Experimental Standards, Reagents and Materials
Six UV-filters: benzophenone (BP), 2-ethylhexyl salicylate (EHS), benzyl salicylate (BS), benzophenone-3 (BP3), menthyl anthranilate (MA), and 2-ethylhexyl 4-(dimethylamino)benzoate (OD-PABA); one disinfectant: triclosan (TCS); and one insect repellent: N,N-diethyl-m-toluamide (DEET), were studied. All standards presented purity higher than 98%, being supplied by Sigma-Aldrich (Steinheim, Germany). Individual stock solutions were prepared in cyclohexane (2040–2590 mg/L), or in acetonitrile (1040-1580 mg/L), depending on the experiment. A mix of PCPs as an intermediate standard solution was prepared daily by dilution of the original individual stock solutions, using cyclohexane or acetonitrile as solvents (also depending on the specific experiment). Working aqueous standard solutions were prepared daily by dilution of intermediate solutions in acetonitrile. All solutions were stored at 4 °C, and protected from light.
Ultrapure deionized water was obtained using the Milli-Q gradient A10 water purification system from Millipore (Watford, UK). Methanol and cyclohexane were purchased from Honeywell (Seelze, Germany), ethyl acetate (EA) from Panreac (Barcelona, Spain), and acetonitrile from VWR International (Barcelona, Spain). Glacial acetic acid was acquired from Millipore (Darmstadt, Germany), and anhydrous sodium acetate (>99%) from Sigma-Aldrich. Both were used in the preparation of an acetic–acetate buffer solution.
The following reagents were used in the synthesis of the CIM-81 MOF: 1,2,4-triazol (98%) and benzene-1,4-dicaboxilic acid (98%), from Sigma-Aldrich; zinc nitrate hexahydrate (98%), from Honeywell; and N,N-dimethylacetamide (DMA), from Merck. The synthesis of the CIM-81 MOF was carried out using 45 mL teflon solvothermal reactors and stainless steel autoclaves, supplied by Parr Instrument Company (Moline, IL, USA).
Pyrex centrifuge tubes (Staffordshire, UK), a 2-mL glass syringe Fortuna Optima (Sigma-Aldrich), and 0.2-µm PVDF (polyvinylidene fluoride) Whatman syringe filters (GE Healthcare, Buckinghamshire, UK), were used in the microextraction procedure.
Sample Collection
Two bottled waters (acquired in local shops) and tap water (collected in the laboratory, and analyzed the same day of the sampling) were used in the analyses.
Instrumentation
The chromatographic analysis was performed on a 7820A gas chromatograph from Agilent Technologies (Waldbronn, Germany), equipped with a HP-5ms ultra inert capillary column (30 m × 0.25 mm × 0.25 µm), and coupled to an Agilent 5977B mass spectrometer equipped electronic impact ionizer and a simple quadrupole as mass analyzer.
A Reax Top vortex of Heidolph (Schwabach, Germany), a centrifuge from Eppendorf model 5720 (Hamburg, Germany), and an IKA RV10 rotary evaporator with a CVR 3000 vacuum controller supplied by VWR, were used in the extraction procedure.
The oven used in the synthesis of the CIM-81 MOF was the Universal UF30 model, supplied by Memmert (Schwabach, Germany).
The phase identification of MOF crystals was performed with an X'Pert diffractometer, supplied by PANalytical (Eindhoven, Netherlands), operating with Bragg-Brentano geometry. Data collection was carried out using Cu K1 radiation (λ = 1.5418 Å) over the angular range from 5.01° to 80.00° (0.02° steps), with a total exposure time of 30 min. IR spectra (450-4000 cm-1) were recorded for the powdered crystals using an IRAffinity1 spectrophotometer from Shimadzu (Kyoto, Japan), equipped with a Pike technologies GladiATR. A Gemini V 2365 model analyzer, supplied by Micromeritics (Georgia, USA), was used to measure the nitrogen adsorption isotherms, in the range 0.02 ≤ P/P0 ≤ 1.00 at 77 K. The Brunauer, Emmet, and Teller (BET) method was used to calculate the surface area. Thermogravimetric analysis on freshly crushed crystals of CIM-81 was carried out in a PerkinElmer Pyris Diamond TG/DTA thermal analyzer (Billerica, MA, USA), requiring a few milligrams under a nitrogen atmosphere, and at a flow rate of 20 mL/min. The temperature was ramped from 25 to 250 °C at a heating rate of 5 °C/min.
Procedures
Synthesis of the MOF
The MOF [Zn2(tz)2(bdc)]·2DMA, termed CIM-81, was synthesized according to a previous method developed by our group (36). Briefly, Zn(NO3)2·6H2O (592 mg, 2 mmol), 1,2,4-triazole (140 mg, 2 mmol), and benzene-1,4-dicarboxylic acid (170 mg, 1 mmol), were dissolved in 15 mL of DMA, and then placed in a Parr Teflon-lined stainless steel vessel (25 mL) under autogenous pressure. The synthesis was carried out at 120 °C for 72 h. The obtained colorless crystals were filtered, washed with DMA and acetone, and dried at 50 °C. The obtained MOF was activated by immersion in acetone for 24 h, to ensure removal of the DMA solvent. The obtained yield was 83%. The crystalline structure was ensured by comparing the experimental X-ray diffraction pattern obtained with the theoretical pattern, thus confirming the nature of CIM-81 MOF already reported by our research group (36).
GC–MS Method
The optimal oven program for the GC separation of the PCPs starts at 70 °C, and increases at a rate of 15 °C/min up to 300 °C, being then held for 5 min. Helium was used as carrier gas at 1 mL/min. The temperature of the transfer line was set to 290 °C, 230 °C for the ionization source, and 150 °C for the quadrupole. The ionization by electronic impact (EI) was carried out at 70 eV. The MS ions used for the monitoring of PCPs are summarized in Table I. The volume of the extract (or standard) injected in the GC–MS was 2 µL in splitless mode at 280 °C, and with a solvent delay of 7.5 min.


D-µSPE Method
The optimal microextraction method requires the addition of 10 mg of the CIM-81 MOF to 10 mL of the aqueous sample (containing or not containing PCPs) or to the aqueous standard, at pH 5 adjusted with an acetic–acetate buffer solution. The tube is then subjected to vortex agitation for 1 min, and then centrifuged (2504 g) for 3 min. Afterwards, the aqueous supernatant is removed, and 1.2 mL of methanol is added to perform the desorption of the PCPs trapped by the MOF. Desorption is facilitated by the application of vortex agitation for 4 min. Then, the desorption solvent containing the target analytes is filtered (0.2 µm), and evaporated under vacuum, followed by reconstitution in 100 µL of a mixture of cychlohexane:ethyl acetate with a ratio of 9:1 (v/v).
Results and Discussion GC–MS Method
The optimal chromatographic separation of the eight PCPs took less than 14 min under the conditions described above. Several quality analytical parameters of the GC–MS method are summarized in Table II, obtained with seven calibration levels. For all PCPs, the relative standard deviation (RSD, in %) for the retention times was < 0.07% (n = 40). For all calibrations, the determination coefficients were >0.995.


Limits of detection (LOD) and limits of quantification (LOQ) were calculated as the concentration providing 3 and 10 times, respectively, the standard deviation of the signal generated by a low concentration standard. Thus, the obtained LOQs ranged from 0.4 µg/L for menthyl anthranilate to 4.6 µg/L for triclosan, as can be seen in Table II.
The precision of the GC–MS method was evaluated at two levels of concentration: low (15 µg/L) and intermediate (45 µg/L), and in intraday (n = 4) and interday with three different non-consecutive days (n = 12). For the lower concentration level, intraday RSD values ranged between 2.6 and 9.7% for benzyl salicylate and benzophenone-3 respectively, while interday values <15% were obtained in all cases. For the intermediate concentration level, values below 4.8 and 9.4% were obtained for the intraday and the interday precision, respectively. Figure 1(a) shows a representative chromatogram of a standard at a concentration of 10 µg/L.


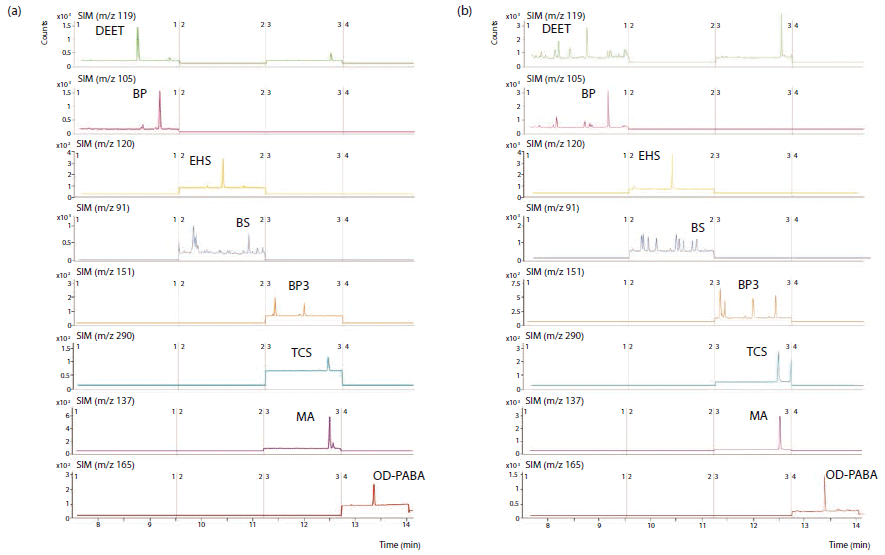
Figure 1: Representative chromatogram of (a) standard solutions of personal care products (PCPs) at a concentration of 10 µg/L, and (b) sample 2 spiked with the PCPs at 4 µg/L concentration and subjected to the entire dispersive mode miniaturized solid-phase extraction-gas chromatography–mass spectrometry (D-µSPE-GC-MS) method. Key to abbreviations: DEET = N,N-diethyl-m-toluamide, BP = benzophenone, EHS = 2-ethylhexyl salicylate, BS = benzyl salicylate, BP3 = benzophenone-3, TCS = triclosan, MA = menthyl anthranilate, OD-PABA = 2-ethylhexyl 4-(dimethylamino)benzoate.
Optimization of the D-µSPE Procedure Combined with GC–MS
The CIM-81 MOF has been used successfully in our research group as sorbent in a D-µSPE method for a group of PCPs (with only 3 PCPs in common with the current study), when the extraction procedure was combined with UHPLC-UV (36). In fact, the majority of publications for MOFs in D-µSPE are in combination with LC (12,28,29). Clearly, the adaptation of methods using MOFs in D-µSPE with GC would make it possible to monitor semivolatile PCPs. In this particularly case, CIM-81 in D-µSPE, the group of PCPs was extended to incorporate DEET, 2-ethylhexyl salicylate, benzyl salicylate, menthyl anthranilate , and 2-ethylhexyl 4-(dimethylamino)benzoate, which were not previously studied (36).
For the purpose of simplifying the adaptation of LC methods with MOFs to GC methods, the main variables of the microextraction procedure were kept constant. Solvent compatibility is certainly crucial for ensuring proper adaptation of the D-µSPE method to GC–MS. Therefore, the first study evaluated the effects caused by changing the desorption solvent of the D-µSPE method (methanol) to a solvent (such as cychlohexane) compatible with the GC column used. The experiments required the addition of 10 mg of the CIM-81 MOF to 10 mL of the aqueous standard containing PCPs at 5 µg/L (and at pH 5 adjusted with an acetic–acetate buffer solution). After vortex agitation (1 min) and centrifugation (2504 g for 3 min) of the tube, the aqueous supernatant was removed, and 1.2 mL of desorption solvent (cychlohexane) is added to perform the desorption step of the PCPs. Desorption was facilitated by the application of vortex agitation for 4 min. Then, cychlohexane, as the desorption solvent, was filtered (0.2 µm), and injected directly in the GC system. Experiments were performed in triplicate.
It was observed that the extraction efficiencies (ER %) obtained for PCPs were not higher than 7%, except for menthyl anthranilate (ER ~20%) and 2-ethylhexyl 4-(dimethylamino)benzoate (ER ~33%). The selection of cychlohexane as the desorption solvent, while adequate for the GC column, was not beneficial in the desorption of PCPs already trapped by the CIM-81 after D-µSPE.
Considering these results, methanol was kept as the desorption solvent in D-µSPE with CIM-81, and therefore a solvent-exchange step was considered. The experiments were carried out using 1.2 mL of methanol as desorption solvent, but followed by an additional step of evaporation and reconstitution. Thus, the methanol was filtered (0.2 µm), and evaporated under vacuum (~8 min), followed by reconstitution in 100 µL of cychlohexane. This volume was the minimum required to ensure proper mixing of PCPs in cychlohexane within the round-bottom flask of the rotary evaporator. As shown in Figure 2, the ER values obtained for most PCPs improved significantly, reaching values up to 40%, thus supporting the validity of this simple approach. It must be considered that ER values close to 100% are rarely obtained in microextraction methods (37), and lower values are acceptable, as long as the sensitivity and precision meet the desired requirements.


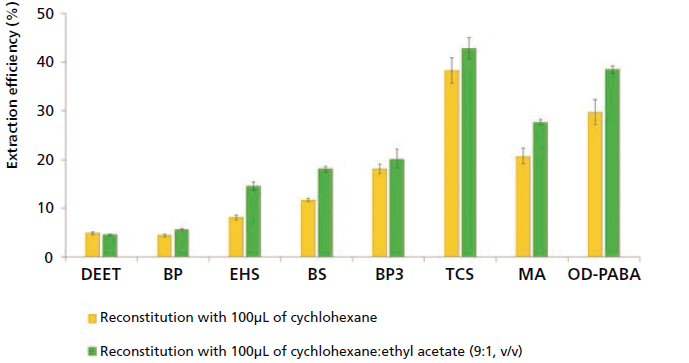
Figure 2: Evaluation of the influence of the volume of the reconstitution solvent: 100 µL (yellow bars), together with the evaluation of the influence of the solvent polarity in the final reconstitution step (green bars). Experiments were carried out in triplicate. Key to abbreviations: DEET = N,N-diethyl-m-toluamide, BP = benzophenone, EHS = 2-ethylhexyl salicylate, BS = benzyl salicylate, BP3 = benzophenone-3, TCS = triclosan, MA = menthyl anthranilate, OD-PABA = 2-ethylhexyl 4-(dimethylamino)benzoate.
Once the adequacy of the solvent-exchange step was shown, while keeping the optimum values of performance for the D-µSPE method, the next step was to test whether higher volumes of cychlohexane were required in the reconstitution, to ensure proper solvation of PCPs in the rotary evaporator glass ball. Thus, 200 µL of cychlohexane was tried, but without clear improvements. Therefore, 100 µL was taken as adequate. Lower values were not tried, because they were clearly insufficient with the 2 mL capacity of the rotary evaporator glass ball.
Ultimately, a slight increase in the polarity of the reconstitution solvent was considered to aid in the solvation of PCPs in cychlohexane. Therefore, a test was done using a miscible solvent (ethyl acetate) along with cychlohexane, which was compatible with the GC column. Specifically, 100 µL of a mixture of cychlohexane and ethyl acetate with the ratio 9:1 (v/v), was evaluated as the reconstitution solvent. Clearly, the mixture provided better ER values than those obtained when reconstituting with 100 µL of cychlohexane, as shown in Figure 2, reaching values up to 43%.
In summary, the selected conditions for the D-µSPE-GC–MS method for the group of semivolatile PCPs were the same as the conditions used in the D-µSPE-UHPLC-UV method (36), but subjecting the methanol desorption solvent (containing trapped PCPs) to filtration, evaporation, and further reconstitution in 100 µL of a mixture of cychlohexane and ethyl acetate with a ratio of 9:1 (v/v).
Analytical Performance of the Optimized D-µSPE-GC–MS Method
Several quality analytical parameters of the proposed D-µSPE-GC–MS method, obtained by subjecting aqueous standards to the entire method, are summarized in Table III. The working range of the calibration curve for all PCPs ranged from 0.2 to 15 µg/L. Calibration parameters were obtained with six concentration levels, and the precision study was carried out at two calibration levels: low (0.8 µg/L), and intermediate (4 µg/L) within the calibration range.


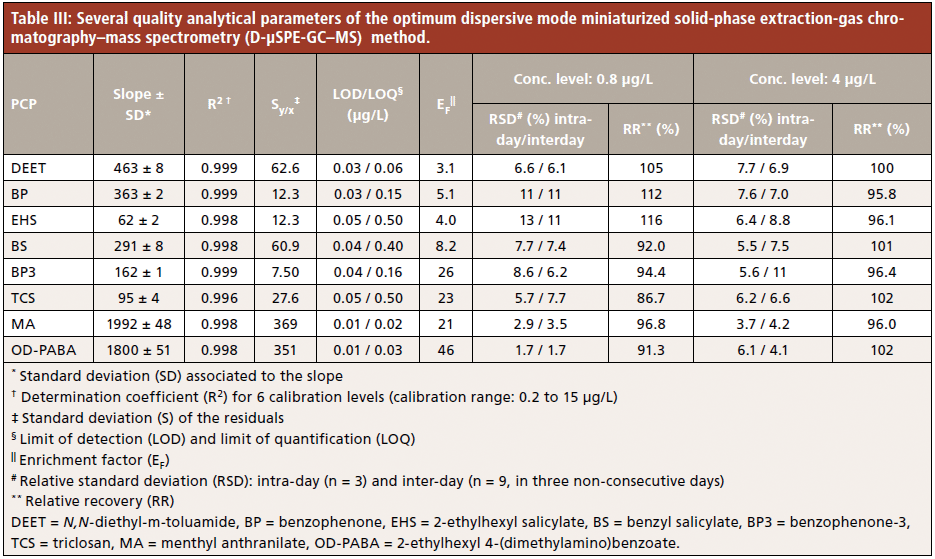
Determination coefficients higher than 0.996 were obtained in all cases. LOD and LOQ were estimated in the same manner as in GC–MS, as the concentration that provided 3 and 10 times, respectively, the standard deviation of the signal given for the final extract of an aqueous standard of low concentration, which was subjected to the entire D-µSPE-GC–MS method. LOD values ranged from 0.01 (menthyl anthranilate and OD-PABA) to 0.05 (2-ethylhexyl salicylate and triclosan) µg/L, and LOQ values ranged between 0.02 (menthyl anthranilate) and 0.50 (2-ethylhexyl salicylate and triclosan) µg/L. These values are around 5 and 30 times lower (depending on the PCP) than those obtained with the GC–MS method, and lower than those described in the UHPLC-UV method for the PCPs in common (benzophenone, benzophenone-3, and triclosan) (36).
The enrichment factor (EF) of the developed method was calculated using an intermediate aqueous standard subjected to the entire method in triplicate, ranging from 3.1 for DEET to 46 for OD-PABA, as shown in Table III, being the maximum (theoretical) EF of 100.
The intermediate precision of the method was evaluated in terms of RSD (in %) for experiments carried out in the same day (intraday, n = 3) and in three different and nonconsecutive days (interday, n = 9, 3 each day), using two concentration levels. For the low concentration level (0.8 µg/L), intraday RSD values were lower than 13%, while interday RSD values were lower than 11%. The relative recoveries (RR) were calculated at both concentration levels, ranging between 86.7% for triclosan and 116% for 2-ethylhexyl salicylate at the low concentration level for triclosan and OD-PABA.
Analysis of samples
The proposed D-µSPE-GC–MS method was applied in the analysis of three samples: two samples of bottled water and one of tap water. Table IV summarizes the results obtained. PCPs were not detected in the bottled water (samples 1 and 2). However, three PCPs were detected in tap water (sample 3). Thus, benzophenone was detected but not quantified, and the quantified PCPs included 5.5±0.9 µg/L of 2-ethylhexyl salicylate and 3.3±0.6 µg/L of triclosan.


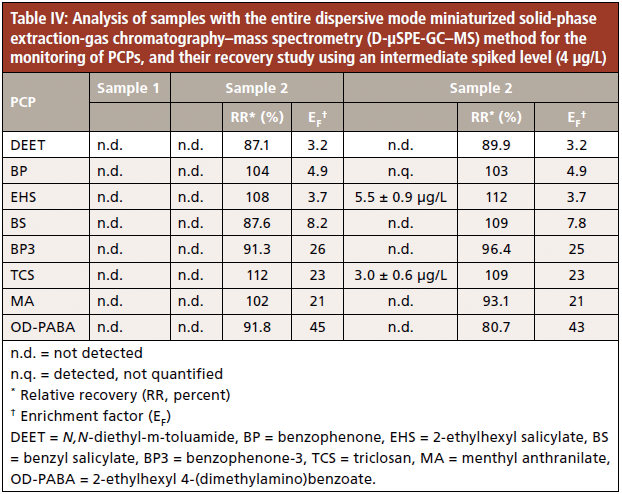
Furthermore, two of the studied samples were spiked to carry out a recovery study to evaluate possible matrix effects. The results obtained for samples 2 and 3 using an intermediate concentration level (4 µg/L) are shown in Table IV. RR values ranged from 87.1% for DEET to 112% for triclosan in sample 2, and from 80.7% for OD-PABA to 112% in sample 3, thus indicating that matrix effects were negligible. Moreover, the obtained EF values for the spiked samples are in agreement with those obtained with the aqueous standards (see Table III), except for OD-PABA.
Figure 1(b) shows a representative chromatogram obtained after the analysis of sample 2 spiked at 4 µg/L and subjected to the entire D-µSPE-GC–MS method.
Conclusions
The promising CIM-81 MOF has been used successfully in a dispersive miniaturized solid-phase extraction method combined with GC–MS to determine a group of personal care products in waters. The resulting method is characterized by being fast (~8 min overall for the entire extraction-desorption step), for requiring low amounts of sorbent (10 mg of CIM-81), for its low sample volume requirements (10 mL of water) and low desorption solvent volumes (1.2 mL of methanol), for ensuring simple compatibility with GC (solvent-exchange step reconstituting with 100 µL of cyclohexane:ethyl acetate, 9:1 ratio), while achieving adequate precision (<13% as interday RSD) and sufficient sensitivity (LODs down to 0.01 µg/L). The reported method ensures a simple adaptation of the D-µSPE approach previously developed for LC, to this method reporting for first time the determination of semivolatile PCPs with GC–MS using D-µSPE with MOFs.
Acknowledgments
P.G.-H. thanks the Agencia Canaria de Investigación, Innovación y Sociedad de la Información (ACIISI), cofunded by the European Social Fund, for her FPI PhD fellowship. Authors acknowledge the beamtime at the BL04–MSPD beamline at ALBA Synchrotron and the collaboration of the ALBA staff (O. Vallcorbá). V.P. acknowledges funding from the Spanish Ministry of Economy (MINECO) project ref. MAT2017-89207-R. A.B.L. thanks the DIAD Group ES for financial support.
References
(1) M. Pedrouzo, F. Borrull, R.M. Marcé, and E. Pocurull, Trends Anal. Chem. 30, 749–760 (2011).
(2) S. Montesdeoca-Esponda, L. Checchini, M. Del Bubba, Z. Sosa-Ferrera, and J.J. Santana-Rodríguez, Sci. Total Environ. 633, 405–425 (2018).
(3) P. González-Hernández, V. Pino, J.H. Ayala, and A.M. Afonso, Anal. Methods 7, 1825–1833 (2015).
(4) R. Suárez, S. Clavijo, J. Avivar, and V. Cerdà, Talanta 148, 589–595 (2016).
(5) I. Pacheco-Fernández, V. Pino, J.H. Ayala, and A.M. Afonso., J. Chromatogr. A 1559, 102–111 (2018).
(6) D. Molins-Delgado, D. García-Sillero, M.S. Díaz-Cruz, and D. Barceló, J. Chromatogr. A 1544, 33–40 (2018).
(7) S.S. Lakade, F. Borrull, K.G. Furton, A. Kabir, R.M. Marcé, and N. Fontanals, Anal. Bioanal. Chem. 410, 2991–3001 (2018).
(8) V. Homem, E. Silva, A. Alves, and L. Santos, Chemosphere 139, 276–287 (2015).
(9) F. Rocha, V. Homem, J. Castro-Jiménez, and N. Ratola, Sci. Total Environ. 650, 2364–2373 (2019).
(10) A. Chisvert, J.L. Benedé, J.L. Anderson, S.A. Pierson, and A. Salvador, Anal. Chim. Acta 983, 130–140 (2017).
(11) S.A. Pierson, M.J. Trujillo-Rodríguez, and J.L. Anderson, J. Sep. Sci. 41, 3081–3088 (2018).
(12) P. Rocío-Bautista, P. González-Hernández, V. Pino, J. Pasán, and A.M. Afonso, Trends Anal. Chem. 90, 114–134 (2017).
(13) I. Pacheco-Fernández, A. Gutiérrez-Serpa, P. Rocío-Bautista, and V. Pino, Solid-Phase Microextraction: Advances in Research and Applications (Nova Science Publishers: New York, USA, 2017), Chapter 4, pp. 147-168.
(14) A. Azzouz, S.K. Kailasa, S.S. Lee, A.J. Rascón, E. Ballesteros, M. Zhang, and K.-H. Kim, Trends Anal. Chem. 108, 347-369 (2018).
(15) M. Varcárcel, S. Cárdenas, B.M. Simonet, Y. Moliner-Martínez, and R. Lucena, Trends Anal. Chem. 27, 34-43 (2008).
(16) R. Lucena, B.M. Simonet, S. Cárdenas, and M. Valcárcel. J. Chromatogr. A 1218, 620-637 (2011).
(17) Y. Wang, M. Rui, and G. Lu, J. Sep. Sci. 41, 180-194 (2018).
(18) O.M. Yaghi, and H. Li, J. Am. Chem. Soc. 117, 10401-10402 (1995).
(19) H. Furukawa, K.E. Cordova, M. O'Keeffe, and O.M. Yaghi, Science 341, 1230444 (2013).
(20) P. Rocío-Bautista, I. Pacheco-Fernández, J. Pasán, and V. Pino, Anal. Chim. Acta 939, 26-41 (2016).
(21) F. Maya, C.P. Cabello, R.M. Frizzarin, J.M. Estela, G.T. Palomino, and V. Cerdà, Trends Anal. Chem. 90, 142-152 (2017).
(22) P. Rocío-Bautista, V. Pino, J.H. Ayala, J. Pasán, C. Ruiz-Pérez, and A.M. Afonso, Talanta 139, 13–20 (2015).
(23) J. González-Sálamo, M.A. González-Curbelo, J. Hernández-Borges, and M.A. Rodríguez-Delgado, Talanta 195, 236-244 (2019).
(24) N. Li, L. Wu, L. Nian, Y. Song, L. Lei, X. Yang, K. Wang, Z. Wang, L. Zhang, H. Zhang, A. Yu, and Z. Zhang, Talanta 142, 43-50 (2015).
(25) P. Rocío-Bautista, V. Pino, J. Pasán, I. López-Hernández, J.H. Ayala, C. Ruiz-Pérez, and A.M. Afonso, Talanta 179, 775–783 (2018).
(26) E. Tahmasebi, M.Y. Masoomi, Y. Yamini, and A. Morsal, Inorg. Chem. 54, 425–433 (2015).
(27) I. Taima-Mancera, P. Rocío-Bautista, J. Pasán, J.H. Ayala, C. Ruiz-Pérez, A.M. Afonso, A.B. Lago, and V. Pino, Molecules 23, 2869 (2018).
(28) I. Pacheco-Fernández, P. González-Hernández, J. Pasán, J.H. Ayala, and V. Pino, Handbook of Smart Materials in Analytical Chemistry (Wiley: Weinheim, Germany, 2019), Chapter 15, pp. 463–502.
(29) P. González-Hernández, A. Gutiérrez-Serpa, P. Rocío-Bautista, J. Pasán, J.H. Ayala, and V. Pino, Eds, Metal Organic Frameworks: Properties and Applications (Central West Publishing: Australia, 2019), IN PRESS.
(30) Z. Huang and H.K. Lee.,Talanta 143, 366–373 (2015).
(31) S. Zhang, W. Yao, J. Yiang, and H. Zhao, J. Chromatogr. A 1452, 18–26 (2016).
(32) N. Lu, X. He, T. Wang, S. Liu, and X. Hou, Microchem. J. 137, 449–455 (2018).
(33) Y. Jia, Y. Zhao, M. Zhao, Z. Wang, X. Chen, and M. Wang, J. Chromatogr. A 1551, 21–28 (2018).
(34) S. Lin, N. Gan, Y. Cao, Y. Chen, and Q. Jiang, J. Chromatogr. A 1446, 34–40 (2016).
(35) A. Nasrollahpour, S.E. Moradi, and M.J. Baniamerian, Food Anal. Methods 10, 2815–2826 (2017).
(36) P. González-Hernández, A.B. Lago, J. Pasán, C. Ruiz-Pérez, J.H. Ayala, A.M. Afonso, and V. Pino, Molecules, 24, 690 (2019).
(37) M.J. Trujillo-Rodríguez, P. Rocío-Bautista, V. Pino, and A.M. Afonso, Trends Anal. Chem. 51, 87–106 (2013).
Providencia González-Hernández, Ana B. Lago, Jorge Pasán, Juan H. Ayala, Ana M. Afonso, and Verónica Pino are with the University of La Laguna, in Tenerife, Spain. Direct correspondence to: veropino@ull.edu.es








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Determining Enhanced Sensitivity to Odors due to Anxiety-Associated Chemosignals with GC
May 8th 2025Based on their hypothesis that smelling anxiety chemosignals can, like visual anxiety induction, lead to an increase in odor sensitivity, a joint study between the University of Erlangen-Nuremberg (Erlangen, Germany) and the Fraunhofer Institute for Process Engineering and Packaging (Freising, Germany) combined behavioral experiments, odor profile analysis by a trained panel, and instrumental analysis of odorants (gas chromatography-olfactometry) and volatiles (gas chromatography-mass spectrometry).


Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.
. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


