Data Integrity Focus, Part IV: Are Raw Data and Complete Data the Same?
LCGC North America
The meaning of the terms raw data and complete data are explored. One term is from EU GMPs and the other is from US GMPs. Do they mean the same thing?
European Union (EU) good manufacturing practice (GMP) in Chapter 4 on Documentation uses the term raw data, but does not define what it means. US GMP uses the term complete data when referring to laboratory records. Confused? We explore the two terms, and ask if they mean the same thing? Could this be regulatory harmonization by different terms?
This is the fourth "Data Integrity Focus" article of a six-part series. The first presented and discussed a data integrity model to present the scope of data integrity and data governance program for an organization (1). The second part discussed data process mapping to identify data integrity gaps in a process involving a chromatography data system (CDS), and looked at ways to remediate them (2). The CDS was operated as a hybrid system, which was the subject of the third article (3). In this part, we look at two regulatory requirements for laboratory records: raw data that is used in EU GMP Chapter 4 (4), and complete data from US GMP 21 CFR 211.194(a) (5), to determine what is meant by these two terms, and ask the title question: Are raw data the same as complete data?
Understanding the Concept of Raw Data
We begin our regulatory ramble with a understanding what raw data means. The problem, from a good manufacturing practice (GMP) perspective, is that raw data is not a GMP term, as we shall see later in this article. The term was first used in a GMP context in the revised EU GMP Chapter 4 on Documentation (4).
EU GMP Chapter 4 and Raw Data
To begin our discussion, we have to obey Cahn's Axiom, which states succinctly: When all else fails, read the regulation, SOP, manual, or instructions.
In the principle of the chapter, we have three references to raw data:
Records: Provide evidence of various actions taken to demonstrate compliance with instructions (activities, events, investigations), and in the case of manufactured batches, a history of each batch of product, including its distribution.
Records include the raw data which is used to generate other records.
For electronic records regulated users should define which data are to be used as raw data.
At least, all data on which quality decisions are based should be defined as raw data.
Let us look at these sentences one by one, and understand what they mean. In the principle, the different document types are presented and explained. Instructions are one of these, and can be an analytical procedure, standard operating procedure (SOP), protocol or study plan, etc. The first sentence states that when an instruction is executed it generates records, which for an analytical laboratory will be a reportable result or a validation report, for example.
The second sentence then states that records include raw data. Therefore, it follows that raw data are a component of a record. Also, according to Chapter 4, raw data can be used to create other records.
Next, we come to the sentence that is not well understood or actioned within most laboratories, that the electronic record set that comprises raw data must be defined. Finally, the fourth sentence begins with the magnificent phrase "at least." This is interpreted in two ways: if you are an inspector, it means this is the minimum, but we would expect more. For those working in the laboratory, it means this is all we are going to do. Then we need to understand, what is a quality decision? Here's a small list:
- Validation of a method
- Results of raw materials, in process control, active pharmaceutical ingredient or finished product testing
- Laboratory investigations and deviations
- Corrective action and preventive action (CAPA) plans and actions
- Change control
As you can see, quality decision covers a wide spectrum, and if an electronic system is used, you should define the raw data for each one.
No EU GMP Definition of Raw Data
However, let us get back to raw data. Do we have any idea what raw data means? Should we look in the glossary of Chapter 4? Let me give you some free advice. Save your time, there isn't one. The term "raw data" is not defined, and is missing in action. The problem is that, having introduced the term, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) expert circle could not agree on a definition, and left it up to industry to resolve. Given the glacial speed of decision making, most companies have done nothing. So how can this be resolved?
MHRA GXP Data Integrity Guidance
To try and resolve the lack of raw data definition, the UK regulator, the Medicines and Healthcare products Regulatory Agency (MHRA) defined the term in their 2018 GXP Data Integrity guidance document (6) in section 6.2.:
Definition: Raw data is defined as the original record (data) which can be described as the first-capture of information, whether recorded on paper or electronically.
Information that is originally captured in a dynamic state should remain available in that state.
Explanation: Raw data must permit full reconstruction of the activities. Where this has been captured in a dynamic state and generated electronically, paper copies cannot be considered as 'raw data'.
According to the MHRA definition, raw data are the first capture of information, regardless of the medium used for the acquisition. This is the definition provided by the MHRA, but there is one small problem. It's wrong!
To be fair to the Agency, in the explanation it states that raw data must permit the full reconstruction of the regulated work. The main problem is that the definition and explanation are separated, and, as I have seen in both training materials and web posts it is the definition that gets the attention and the explanation is ignored.
What do we do?
US and Organization for Economic Cooperation and Development (OECD) GLP Regulations
You may wonder why we are looking at good laboratory practice (GLP) regulations now. The reason is very simple; raw data is a GLP term, and has been in the US regulations since 1978 (7). In 21 CFR 58.3(k), "raw data" is defined as:
Raw data means any laboratory worksheets, records, memoranda, notes, or exact copies thereof, that are the result of original observations,
and activities of a nonclinical laboratory study,
and are necessary for the reconstruction and evaluation of the report of that study.
Let us interpret the regulation section by section, as we did with Chapter 4 earlier.
Raw data means any record medium that captures original observations. Here we can equate original observations with the MHRA's first-capture of information (6). The US definition also has the option for an exact copy of original observations. The classic example of this is where an instrument prints out a record on thermal paper, or uses ink that fades over time, and the printout will not last the retention period. So, the record is copied, the original and the copy are put side by side on the laboratory notebook or official record, and the analyst verifies that the copy is an exact one.
Now we come to the reason why the MHRA definition of raw data is wrong. Raw data is not just original observations, but includes other activities of a GLP study. These other activities will generate more data and records.
Finally, the original observations and activities are necessary for the reconstruction and evaluation of the report of the study. This equates to the MHRA definition and explanation of raw data, but due to the split between the two as discussed above (6), it is close, but no cigar.
At 40 years old, GLP regulation is showing its age, but, instead of a mid-life crisis, the FDA has issued a proposed update to the GLP regulations in August 2016 for industry comment (8). The raw data definition has been modified slightly, but still retains the same meaning. For interest, the OECD GLP definition of raw data is similar (9). If you want more detail on the definition of raw data, please read my December, 2018 "Focus on Quality" column in Spectroscopy (10) that is the culmination of 22 years of writing about the definition of raw data in an electronic world.
What About Raw Data in GMP?
Like a boomerang, we come back to our problem with the definition of raw data in GMP. How will we resolve the issue? Strangely enough, the answer comes from the same MHRA GXP guidance document (6) that did not define raw data correctly. Delve a little further into the document and you'll come to section 6.11 and original record; here the definition states:
- The first or source capture of data or information, such as original paper record of manual observation or electronic raw data file from a computerized system, and
- All subsequent data required to fully reconstruct the conduct of the GXP activity.
Let us undertake an analysis of this definition.
- First or source capture of data (equating to original observations in US GLP) √
- And all subsequent data (equating to and activities) √
- To fully reconstruct the GXP activity (equating to reconstruction and evaluation of the report) √
Three matches and a jackpot! Raw data is everything you acquire or transform between the start and report of a GLP or GMP activity. All that needs to happen is that the names of the definitions are swapped (raw data becomes original record and vice versa), and all is good with the world.
Understanding Complete Data
We have reached half way in our discussion and we now need to consider what are complete data. To do this, we have to cross the pond and look at US GMP.
Able Laboratories 483 Observations
Connoisseurs of FDA warning letters and 483 observations for laboratory inspections will be very familiar with the citation under 21 CFR 211.194(a), such as that for Able Laboratories in July 2005 (11):
Laboratory records do not include complete data derived from all tests, examinations, and assay necessary to assure compliance with established specifications and standards.
The QC Laboratory notebooks and binders lacked data from all analytical testing conducted in the QC Laboratory. Laboratory records did not include all data, such as out of specification (OOS) results, chromatograms, sample weights, and processing methods. OOS results were substituted with passing results by Analysts and Supervisors. The substitution of data was performed by cutting and pasting of chromatograms, substituting vials, changing sample weights, and changing processing methods.
You may wonder why I have put a citation here. The rationale is that this is THE 483 observation that triggered the data integrity tsunami from the FDA, and then other regulatory agencies. If the first result did not pass, then Able falsified the data into compliance.
In contrast to the limbo dancing for raw data, complete data is relatively easy to understand:
21 CFR 211.194(a) Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays as follows (5)
The two word phrase "complete data" is self-explanatory: everything captured or generated during the course of an analysis. The undocumented corollary is that nothing is left out, ignored, swept under the carpet, or deleted.


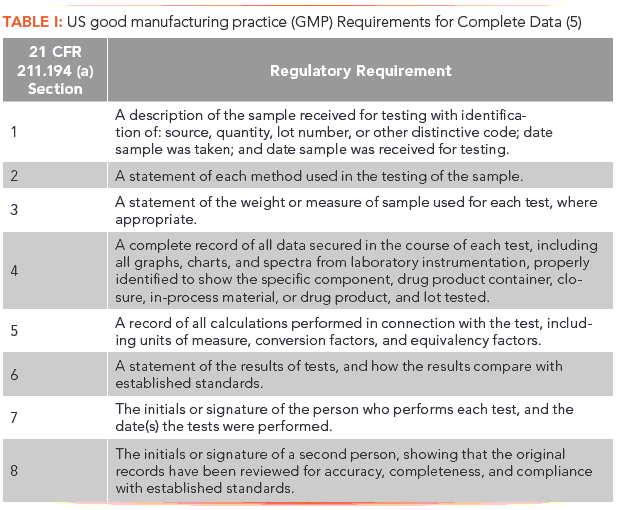
There clause is completed with a list of eight items, as shown in Table I, that are required to ensure complete data. Of interest to our discussion is sub-clause 4: A complete record of all data secured in the course of each test (5). See? It's that word "complete" again, and, coupled with "all data" means that an organization cannot be selective with the data and records it keeps. Complete means complete even if an analysis has:
- An aborted run
- Failed system suitability tests
- An instrument failure
- An OOS result that may require reinjection of extracts as part of an investigation
All data must be collected and retained. Moreover, from sample to reportable result, there must be a traceable data set, and, if data are excluded, there must be a scientifically sound rationale (21 CFR 211.160(b) [5]).
Out of interest, 21 CFR 211.188 requires "complete information" for production records (5). The reason for this is that production data are mostly static (for example, weights and temperatures) that a person cannot interact with and interpret. In contrast, "complete data" for laboratory records (such as chromatograms and spectra) need to be interpreted, and are typically dynamic data.
Are Raw Data and Complete Data the Same?
Now we are ready to answer the question posed in the title: Are raw data and complete data the same? What do you think? Let me give you a clue– the only answers are "Yes," or "No." Unfortunately for you, there is no audience to ask, and I don't know if you have phoned a friend. Let us review the definitions and explanations:
Raw data: original observations and activities of a study and are necessary for the reconstruction and evaluation of the report
Complete data: all data from sampling to generation of the reportable result including the second person review
To all intents and purposes, raw data and complete data are the same.
Practical Interpretation for Chromatography
OK, if the two terms are equivalent, what does this mean in practice for a chromatographic analysis? If we return to the MHRA GXP data integrity guidance (6), there is a discussion about data and metadata. A value of 98.3 is meaningless on its own. What does this mean? The MHRA guidance document lays out an explanation for a simple situation. Guidance is, of necessity, high level, and leaves the reader to interpret it to their own situation.
Chromatographic analysis is more complex than the example given in the MHRA guidance document. My interpretation of complete data and raw data for chromatographic analysis is shown in Figure 1.

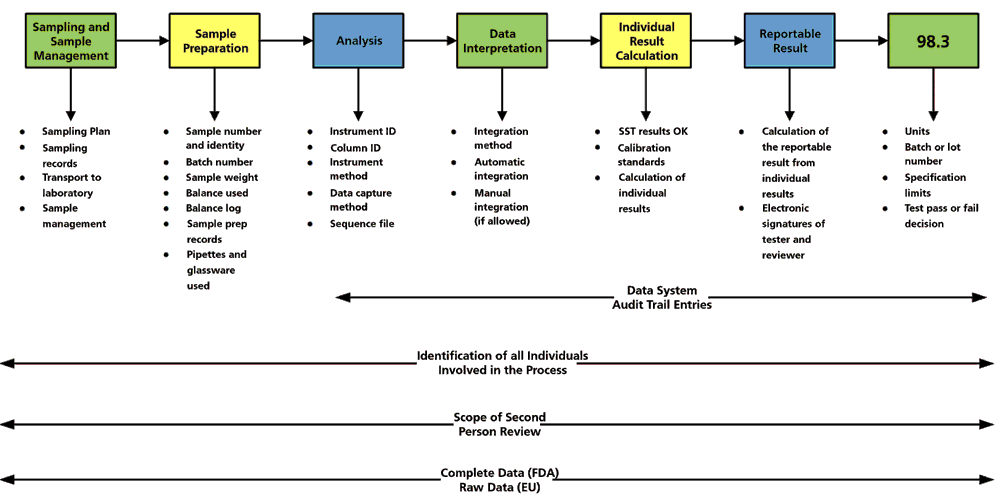
Figure 1: Raw Data and Complete Data for a Chromatographic Analysis (12).
Here the full scope of the analysis from taking the sample through analysis, interpretation to calculation of the reportable result can be seen along with the major data elements. Depending on the processes and computerized systems in place there will be paper as well as electronic records throughout the workflow.
Summary
We have looked at EU GMP Chapter 4, and interpreted the term raw data by using the definition from US GLP regulations. We have also understood what complete data mean in a GMP laboratory. We can demonstrate that raw data and complete data are equivalent, and show what this means in practice for a chromatographic analysis.
References
(1) R.D. McDowall, LCGC N. Amer. 37(1), 44–51 (2019).
(2) R.D. McDowall, LCGC N. Amer. 37(2), 118–123 (2019).
(3) R.D. McDowall, LCGC N. Amer. 37(3), 180–184 (2019).
(4) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation (E. Commission, Editor, Brussels, Belgium, 2011).
(5) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Sliver Springs, Maryland, 2008).
(6) MHRA GXP Data Integrity Guidance and Definitions. Medicines and Healthcare products (Regulatory Agency, London, United Kingdom, 2018).
(7) 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies. (Food and Drug Administration: Washington, D.C., 1978).
(8) 21 CFR Parts 16 and 58 Good Laboratory Practice for Nonclinical laboratory Studies; Proposed Rule. Federal Register, 2016. 81(164), 58342–58380.
(9) OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice. Organsation for Economic Co-operation and Development, Paris, France, 1998).
(10) R.D.McDowall, Spectroscopy, 2018. 33(12), 8–11 (2018).
(11) Able Laboratories Form 483 Observations. 2005 1 Jan 2016]; Available from: http://www.fda.gov/downloads/aboutfda/centersoffices/officeofglobalregulatoryoperationsandpolicy/ora/oraelectronicreadingroom/ucm061818.pdf.
(12) R.D.McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com













Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


The Effect of Time and Tide On PFAS Concentrations in Estuaries
April 8th 2025Oliver Jones and Navneet Singh from RMIT University, Melbourne, Australia discuss a recent study they conducted to investigate the relationship between tidal cycles and PFAS concentrations in estuarine systems, and offer practical advice on the sample preparation and LC–MS/MS techniques they used to achieve the best results.



