Data Integrity and USP , Part 2: OQ Supervision and Execution
LCGC Europe


This is the second of three articles looking at the impact of the new United States Pharmacopeia (USP) on analytical instrument qualification (AIQ) on data integrity in a regulated chromatography laboratory. This part focuses on how the laboratory should supervise the execution of operational qualification (OQ) protocols by a third-party service provider. The principles described also apply to in-house metrology departments.
This is the second of three articles looking at the impact of the new United States Pharmacopeia (USP) <1058> on analytical instrument qualification (AIQ) on data integrity in a regulated chromatography laboratory. This part focuses on how the laboratory should supervise the execution of operational qualification (OQ) protocols by a third-party service provider. The principles described also apply to in-house metrology departments.
The first part of this three-part series focused on the impact of the new version of the United States Pharmacopeia (USP) <1058> (1) and looked at instrument specifications and the role of suppliers (2). As demonstrated in part 1, analytical instrument qualification (AIQ) is level 1 of the data integrity model (2–4) and is therefore an essential component for ensuring data integrity.
The Qualification Continuum
Before going into detail about operational qualification (OQ), it is important to understand that AIQ is a dynamic continuum that spans all instrument stages associated with:
- Specification, purchase, and implementation
- Ongoing qualification
- Routine use.
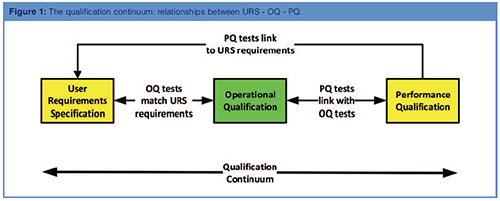
A key component of this continuum is the relationship between the user requirement specification (URS), OQ, and performance qualification (PQ) as shown in Figure 1. The new USP <1058> (1) states that users must write a URS for their instrument (yes, even for a pH meter) and this must be tested in the OQ. What is equally important, and will be covered in Part 3, is that the PQ must also demonstrate that the instrument meets intended use requirements during the operational phase. Therefore, as tests for the OQ are developed, consideration should also be given to the plans and tests that need to be performed during the PQ phase to demonstrate conformance with URS requirements.
- Will any OQ tests be repeated in the PQ?
- Are new tests required for the PQ?
- What is different and why?


For chromatography instruments, many laboratories have historically applied a life cycle process based on a minimum sub-set of the life cycle in the 2008 version of USP <1058> (5):
- Supplier: Responsible for IQ/OQ, preventative maintenance, and an annual OQ
- Laboratory: Responsible for system suitability tests (SST) and in some mindsets SST = PQ
Limitations of this segmented approach include the fact that the AIQ is disconnected from routine use or applications and components of the full life cycle are missing. There is no laboratory URS and requirements are informally based on pharmacopoeial general chapters. Diagnostic tests performed by the instrument are another important component of assuring instrument functionality, for example, when a UV–vis high performance liquid chromatography (HPLC) detector is powered up a wavelength test based on emission lines from the lamp is performed.
A Quick Recap
Historically, when process validation principles were first applied to laboratory instruments, there was an over reliance on documentation (6) and many of the life cycle steps, such as URS and design qualification (DQ), were considered clerical steps that were the responsibility of the supplier and potentially independent of how the instrument would be used in an individual laboratory. This is wrong (7). In Part 1 (2), we discussed why users must write their own specifications for the analytical instruments that they purchase based on their intended use, and not blindly use those from a supplier.
This means that the URS has evolved from what was a tick box activity to becoming the most important document in the AIQ life cycle.
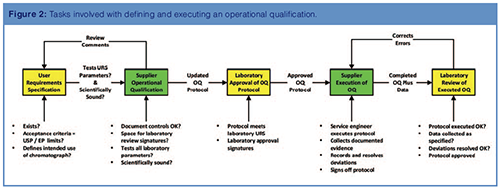
USP <1058> requires that suppliers must publish meaningful specifications relating to instrument parameters that users can test practically in their own laboratories, rather than in a controlled environment. In addition, the URS for a chromatograph must be the basis for intended use testing in the OQ phase (1). In this part, we will address the writing, execution, supervision, and execution of OQ tests as shown in Figure 2.

Merging IQ and OQ?
There is harmonization between the new USP <1058> and EU GMP Annex 15 on Qualification and Validation when it comes to merging qualification documents because both allow this (8). The main documents that can be merged are typically the IQ and OQ protocols. This has advantages for both the laboratory and the supplier because there is a single document to prepare, review, and execute. It also permits a service engineer to move from the IQ to the OQ without having to wait for a laboratory approval, thereby allowing for more efficient execution.
Quality by Design or Compliance by Stupidity?
When designing OQ tests it is important to understand the following factors:
- How an instrument and any associated software works;
- Know the applicable pharmacopoeial and regulatory requirements;
- The application of sound science and risk-based qualification
Let us explain what we mean. How would you measure flow rate accuracy and precision of an HPLC pump? Table 1 shows an example of how to measure pump flow rate (9).

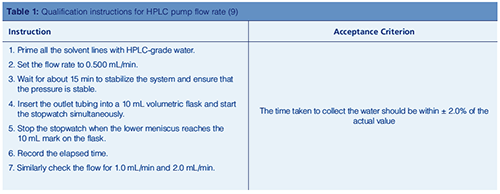
Let us analyze this approach. Is it scientifically sound? No!
- Where is the user requirement for flow rate and acceptance criteria? The paper does not mention the instrument specification
- Flow rate accuracy is measured but there is only one measurement, therefore flow rate precision cannot be determined
- Is the stopwatch calibrated? Probably not, as it does not say.
- Is there a column attached to the pump? The paper does not say, but if not then the measurement does not cover intended use.
Compliance by stupidity? Yes!
- The normal qualification approach would be to measure the lower
- and upper limit of flow rate. Interpolation is acceptable, extrapolation is not.
- In the example, an additional middle flow rate is added “just in case”.
- There is no objective evidence of the measurement, only a single observation by a person. Yet, the laboratory has agreed to this approach.
Instead, simplify the process by purchasing a calibrated digital flow meter and conduct six measurements at the top and bottom flow rates of the user specification. This is faster and more accurate than shown above and if the flow meter is linked to a printer or laboratory informatics application there is objective evidence of the test as well as better data integrity.
Our second example is a UV diode array HPLC detector. Apart from the initial wavelength check during the first OQ, why would you want to check the wavelength again? This last sentence may raise some eyebrows. Why would you not check wavelength accuracy? Remember, know your instrument: the diode array is superglued to the optical bench. It does not move. Therefore, why check the wavelength annually? Most chromatographers will take the path of least resistance and perform an unnecessary qualification because it is easier to defend than take a justified risk-based approach.
Tests performed on a chromatography instrument during qualification typically fall into the following categories:
- Metrology performance (for example, flow, temperature, pressure)
- Checks versus reference materials (wavelength accuracy)
- Detector noise evaluation
- Injection-based tests (for example, injector precision, accuracy, linearity, and carryover)
- Usage-based tests (typically requires an injection to “trigger” another measurement such as gradient formation but are not specific to the injector)
The URS becoming the most important life cycle document will challenge many laboratories current interpretation of AIQ and USP <1058> (based on how the 2008 version was implemented where instrument specification was the responsibility of the supplier). In the absence of a strong industry or regulatory driver to harmonize AIQ, many laboratories have implemented a diverse interpretation of USP <1058> requirements. Although there is variation from laboratory to laboratory, most quality control (QC) laboratories have a clear understanding of <1058> requirements and what should be included in an OQ and will typically be comfortable with testing the instrument range of use based on current applications.
In contrast, R&D laboratories are more likely to base requirements on the instrument specification, some of which cannot be measured effectively outside of controlled conditions. Thermal drift of the instrument environment contributes significantly to detector drift measurements for an HPLC system. This is why “factory testing” of the instrument occurs in a controlled environment and is a scientific requirement to test the instrument noise and drift specification. However, this causes a challenge when performing detector OQ tests if the laboratory tries to test the instrument performance to the instrument specification, because the laboratory temperature control is not as good as the controlled environment used at the factory. This is an example of where trying to test an instrument to its specification may be less than scientifically valid.
Do I Need a Contract?
EU GMP Chapter 7 is focused on outsourcing. Using a third party such as the supplier or a service agent is outsourcing and as required by Chapter 7 (10):
7.1. There should be a written Contract covering the outsourced activities, the products or operations to which they are related, and any technical arrangements made in connection with it.
A contract or agreement outlining the roles and responsibilities between the laboratory and the supplier is essential for any qualification work. However, what if you use an internal supplier such as an in-house metrology department? We suggest that the approach offered by EU GMP Annex 11 clause 3.1 (11) is appropriate here: there needs to be an agreement with third-party suppliers and the same applies to IT departments. An agreement should also apply to in-house service groups performing qualification services. Do you have a quality or technical agreement with your metrology department?
OQ Tests Must Reflect the Laboratory URS
The new version of USP <1058> makes the following statements related to OQ:
OQ demonstrates fitness for the selected use, and should reflect URS.
Holistic tests, which involve the entire system, demonstrate that the whole system complies with URS (1).
Therefore, the principle for any qualification is that experiments should be designed to reflect the way that you work and that are documented in an instrument URS.
However, the big problem is that most laboratories do not have a user requirements specification for their instruments. Many chromatographers don’t see the need for an instrument specification, but definition of intended use is a requirement of all GXP regulations (12–15). Why can’t you use the supplier’s specification? Our response is read Part 1 of this series (2) and then go and write one.
The URS is an essential requirement for input into the OQ protocol; without one you cannot qualify an analytical instrument. Many laboratories just use a supplier’s OQ protocol and ignore the first two stages shown on the left of Figure 2.
Review and Approval of OQ Protocols
The new USP <1058> states:
For OQ test packages purchased from a service provider or supplier, the user must review the material to assure themselves of the scientific soundness of the tests and compliance with applicable regulations (1).
There are several points to consider that arise from this sentence (Figure 2):
- Are the document controls of the OQ adequate?
- Do the tests match your URS requirements?
- If not, can the supplier configure the OQ to match your requirements (such as adding additional tests and set points) or will the laboratory have to conduct additional qualification experiments?
- How will any “gaps” be managed?
- Are the acceptance criteria set to limits defined in a pharmacopoeial general chapter?
- Is each test in the OQ scientifically sound, for example, is it conducted with calibrated instruments, traceable reference standards, etc?
- How is documented evidence of each test collected and managed?
- Does the laboratory have the electronic records of the testing available for later review?
- Is there space for the laboratory to sign off the final OQ protocol prior to execution?
- Does the laboratory’s organization require a quality assurance (QA) signature prior to execution?
Why? See our discussion later in this column.
Most qualification protocols are paper-based documents and, as such, will typically include instructions for performing the test, spaces to write in the results, tables for documenting deviations (where deviations from the protocol are documented), and a list of attachments. A paper OQ protocol is almost a “stand-alone” quality system. In contrast, as laboratories consider implementation of electronic protocols, the content and format of the protocol does not need to be the same as the paper-based approach (for example, the instructions will be in the application used to execute the qualification and generate the report).
Laboratory Training and Understanding
The laboratory is always responsible for the quality, data integrity, and scientific validity of any AIQ work performed, irrespective of who performs the work (1,14). Historically, where the instrument manufacturer performs an OQ, many laboratories may have approved OQ protocols without a thorough understanding of the test design and how the work will be performed. This “manufacturer knows best” interpretation of <1058> requirements can result in problems during a regulatory audit or inspection-the highest risk a laboratory can take during an audit is to try and explain something they don’t understand! The change in FDA reporting practices means it is not unusual for software vendors and instrument manufacturers to be named in an FDA warning letter or 483 report. This means that laboratories who approve documents without understanding the content represent the greatest risk to the perceived reputation of the supplier. Historical laboratory audits and defence processes (pre-2010) tended to concentrate on the scientific validity of the information. In data integrity focused audits, providing evidence that the results of the OQ are not fraudulent represents a very different kind of audit.
This means that companies performing OQ testing should provide training to laboratories on the OQ protocol, including how to review the electronic information and associated metadata from the OQ. Some OQ tests are performed manually, typically those based on metrology measurements, and currently this can be interpreted as a data integrity risk. Until secure, fully electronic measurement tools are developed (for example, for an HPLC flow meter), there are no other ways the work can be performed. The options available to the laboratory include second person observation of the test being performed. When performing laboratory analysis, sample preparation is one of the most critical steps. Should all sample preparation be observed by a second person?
Where there is an OQ test failure, this can represent a high focus area for an auditor. Because of this, it is not unusual for some laboratories to request that the OQ qualification report does not contain any deviations (by implication, if there is a test failure, don’t include it in the report!). How often an OQ test fails will depend on how tight the OQ specification is for the test. If a laboratory has implemented very tight limits for a test, the test is more likely to fail, prompting additional work for the laboratory and an investigation into the potential impact of the instrument failure on analytical results generated since the previous OQ. Qualification work must include all results, which is easier to enforce with electronic protocols and supporting software, rather than relying on procedural control (instructions) of paper protocols.
One of the areas that needs to be clearly understood relates to limits defined in pharmacopoeia general chapters and drug testing monographs. Instruments must be capable of meeting the requirements of the pharmacopoeia, but this does not have to be performed during the OQ. This is where the performance qualification (PQ) and the valid role of meaningful SSTs come in as part of the continuum of qualification. For example, injection precision is a requirement that is almost always included in SSTs when performing chromatographic testing. The requirements specific in USP <621> (16) must be satisfied. Injection precision tests are included in OQ protocols, but they are based on the test design of the OQ protocol and associated methodology, not on pharmacopoeia requirements. As well as being scientifically valid and developed through an appropriate quality life cycle process, OQ tests are typically developed by suppliers to test instruments in a time efficient way. A supplier or service provider could test every function or combination of parameters of an instrument during an OQ protocol, but this would significantly increase the duration of the OQ protocol to several days as well as the cost. The qualification continuum extends qualification thinking beyond the OQ and across multiple stages of the life cycle. It is this extended continuum that provides robust assurance of the performance of the instrument.
Review and Approval Signatures
USP <1058> is very explicit in this respect:
The user should review the documents before execution and approve the tests after execution to ensure completeness and accuracy of the completed document and the test data generated (1).
Who in the laboratory is going to review and approve qualification protocols and are they trained?
Technical personnel such as those in the laboratory should review and approve the OQ protocol not QA, who may not have the necessary understanding of the underlying analytical technique. This raises a complication: under European GMP, the qualified person (QP) is legally responsible for the quality of the medicines-should they approve the OQ protocol?
Just as many laboratories have approved OQ protocols without fully understanding the OQ test design and rationale, many have also approved the OQ report without a detailed understanding of the information or electronic review of the data. Many laboratories may have a policy where an external service provider cannot access or use the laboratory’s chromatography data system (CDS). This means that the service provider has to control the instrument using software run from a laptop, which the laboratory does not have control over. Additionally, unplugging the instrument from the network to plug into a laptop possibly running different CDS software to that used in the laboratory is not testing the instrument under actual conditions of use. How can the laboratory review audit trail entries if the computer where the records reside is taken off site at the end of the qualification? It is better to work with service providers and define appropriate CDS access for the service personnel to perform the work.
As can be seen in Figure 2, there is typically iteration to agree the OQ protocol before execution and then post-execution approval.
- Pre-execution review should be simple and efficient, not convoluted.
- If there are changes to be made to the OQ protocol, does the supplier, laboratory, or both change control procedures apply?
- Should a supplier refuse to perform work if the protocol has not been approved?
- Should a risk-based approach be applied to review of qualification e-records or should all records be reviewed?
Not reviewing and approving qualification protocols can seriously damage your compliance reputation as seen in 2000 when Spolana, a Czech active pharmaceutical ingredient (API) supplier, received a warning letter with the following citation (17):
1. Written procedures had not been established for the calibration of analytical instruments and equipment in the Quality Control laboratories used for raw material, finished API and stability testing. Furthermore, calibration data and results provided by an outside contractor were not checked, reviewed and approved by a responsible Q.C. or Q.A. official
2. The <redacted> systems calibrated by an outside contractor did not include verification of the precision (% RSD) of the autoinjector at more than one injection volume, the flow rate below 1 ml/min, or the wavelength accuracy for the wavelength regions used for testing of <redacted>
Although this warning letter is nearly 20 years old, the warning is clear. The laboratory is responsible for the technical content and compliance with regulations of the OQ protocols.
Roles and Responsibilities
The role of the laboratory is defined clearly in USP <1058> (1):
For OQ test packages purchased from a service provider or supplier, the user must review the material to assure themselves of the scientific soundness of the tests and compliance with applicable regulations.
The user should review the documents before execution and approve the tests after execution to ensure completeness and accuracy of the completed document and the test data generated.
The supplier of the OQ protocol has the following responsibilities:
- They must provide base OQ protocols.
- They must clarify roles and responsibilities between the two parties for update, review, execution, and approval of the work.
- Is the protocol static where no changes can be made or dynamic where the testing can be adapted to a laboratory’s specification?
- The engineer should be trained not just in executing the protocol but also in data integrity (ALCOA principles) with certification.
- The engineer must not falsify data and must record all work including deviations and their resolution.
- All AIQ activities should be predefined and contemporaneously documented (1).
- Electronic data and the means to read it must be left with the laboratory if the user’s CDS is not used.
Don’t Forget the Software!
Although this discussion is focused on the qualification of the chromatograph system, it is important that the CDS software is not forgotten. The new USP <1058> (1) integrates qualification of the instrument with validation of the software to reflect what happens in practice with Group C instruments: the instrument needs the software and the software needs the instrument to operate. For the most part, the new USP <1058> is now aligned with GAMP 5 principles (18).
- For the installation of a new system there will be both software and instrument qualification protocols (3,19).
- If the laboratory’s CDS is going to be used to execute the instrument OQ, then create a supplier account where the identity of the engineer executing the test can be identified directly or indirectly. Never use the default account in a CDS for performing qualification.
- A periodic review of the overall system is the equivalent of an instrument preventative maintenance and regular OQ.
Dude, Where’s My Data?
In an ideal world, the instrument should be controlled through the laboratory CDS during OQ protocol execution, with all the chromatographic data generated in a single location and under laboratory control. The data generated during the OQ execution are essential to support a claim that any instrument is qualified. If an organization does not allow a supplier to access the CDS, the OQ protocol will be performed using the engineer’s laptop and the data will reside there-not an ideal solution. A situation could arise that the engineer leaves the laboratory and there are only paper records available-a worse situation still. Where data are provided on a CD at the end of the qualification work, this is still a higher risk than having the data stored and backed up automatically within the laboratory’s IT domain (CDs have to be managed through procedural control and can go “missing”!). Regardless of how an OQ is executed, does the engineer have access to the system clock, the data in the operating system directories, and the recycle bin?
Again, in an ideal world, the protocol should be executed and signed electronically, rather than having a hybrid situation of electronic records in a CDS and signed paper printouts. This is the worst situation.
Summary
In part 2, we have discussed the qualification continuum from URS to OQ and PQ. The OQ protocol must be based on the requirements in the laboratory URS for the chromatograph system. Users must review the protocol prior to execution to check for scientific soundness and coverage vs. their URS. The supplier executes the reviewed protocol and the trained engineer collects the data, completes the protocol, and resolves any deviations. The executed protocol is then collected by an engineer trained both in execution of protocol and ALCOA principles. The user reviews after execution both protocol execution and data collected. The instrument is then released for operational use. In Part 3 we will look at the new requirements for performance qualification.
References
- General Chapter <1058> “Analytical Instrument Qualification” in United States Pharmacopeia 41 (United States Pharmacopeial Rockville, Maryland, USA, 2018).
- P. Smith and R.D. McDowall, LCGC Europe 31(7), 385–389 (2018).
- R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Second Edition, Royal Society of Chemistry, Cambridge, UK, 2017).
- R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, UK, 2018).
- General Chapter <1058> “Analytical Instrument Qualification” in United States Pharmacopeia 31 (United States Pharmacopeial Rockville, Maryland, USA, 2008).
- AAPS White Paper on Analytical Instrument Qualification (American Association of Pharmaceutical Scientists, Arlington, Virginia, USA, 2004).
- R.D. McDowall, Spectroscopy 25(11), 24–31 (2010).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation
- D. Gowrisankar et al., Journal of Biomedical Science and Research 2(2), 89–99 (2010).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 7 Outsourced Activities (European Commission, Brussels, Belgium, 2013).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerised Systems (European Commission, Brussels, Belgium, 2011).
- 21 CFR 58, Good Laboratory Practice for Non-Clinical Laboratory Studies (Food and Drug Administration, Washington, DC, USA, 1978).
- OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice (Organisation for Economic Co-operation and Development, Paris, France, 1998).
- 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Silver Spring, Maryland, USA, 2008).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 3 Premise and Equipment (European Commission, Brussels, Belgium, 2014).
- United States Pharmacopeia General Chapter <621> “Chromatography” (United States Pharmacopoeia Convention, Rockville, Maryland, USA).
- FDA, Warning Letter Spolana (Food and Drug Administration, Rockville, Maryland, USA, 2000).
- L. Vuolo-Schuessler et al., Pharmaceutical Engineering 34(1), 46–56 (2014).
- GAMP Good Practice Guide A Risk Based Approach to GXP Compliant Laboratory Computerised Systems, Second Edition (International Society for Pharmaceutical Engineering, Tampa, Florida, USA, 2012).
Paul Smith is a Global Strategic Compliance Specialist at Agilent Technologies. After initially specializing in spectroscopy and application of chemometrics to spectroscopic data, Paul developed his compliance expertise in a variety of quality and management roles within the 17 years he spent in the pharmaceutical industry. Paul worked as an independent consultant and university lecturer before moving into laboratory compliance consultancy roles.
“Questions of Quality” editor Bob McDowall is Director at R.D. McDowall Ltd., Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the editor-in-chief, Alasdair Matheson, at alasdair.matheson@ubm.com













