Data Integrity and USP : Part 1 Specifications and Suppliers
LCGC Europe


This is the first of three articles looking at the impact of the new United States Pharmacopeia (USP) chapter on Analytical Instrument Qualification (AIQ) on data integrity in a regulated chromatography labora-tory. In part 1, user specifications for chromatography systems and the relationship between users and sup-pliers will be discussed.
This is the first of three articles looking at the impact of the new United States Pharmacopeia (USP) <1058> chapter on Analytical Instrument Qualification (AIQ) on data integrity in a regulated chromatography laboratory. In part 1, user specifications for chromatography systems and the relationship between users and suppliers will be discussed.
In January 2018 (1), this column gave an overview of the main changes contained in the new version of United States Pharmacopeia (USP) general chapter <1058> on Analytical Instrument Qualification (2). AIQ is important as shown by the “critical” EudraGMDP citation:
This included a gross failure of change management, permitting the use of an unqualified HPLC system (3).
In this series of articles, we will go into more detail on specific aspects of the new version of USP <1058>. In the first part, we will look at writing user requirement specifications for a chromatograph and the relationship between the chromatography laboratory and an instrument supplier and how these two factors impact data integrity. The second part will look at the execution and supervision of an operational qualification (OQ) protocol and the third part will look at monitoring instrument performance and requalification of chromatographs.
To understand the importance of AIQ in data integrity the scope of data integrity and data governance within an organization must be understood. To help there is a four-layer data integrity model. The analytical portion of the model is shown in Figure 1; for the full model including production and quality assurance see references 4 and 5.


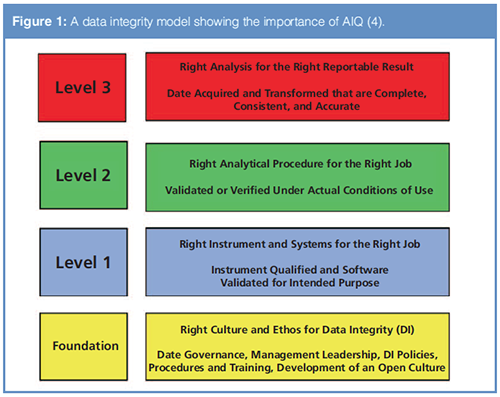
The model consists of four layers and is analogous to building a house where there is a foundation followed by three layers:
- Foundation: Data Governance-the right culture and ethos for data integrity
- Level 1: Analytical Instrument Qualification and Computerized System Validation-the right instrument and computer system for the right job
- Level 2: Analytical Procedure Validation-the right analytical procedure for the right job
- Level 3: Analysis of Samples-application of the levels below to the analysis of samples
The house analogy is fitting because if one of the layers is not present then those above cannot work and the house collapses. This is especially true of the foundation where management leadership is critical for success. From the Data Integrity Model, the first level after the foundation is AIQ and computerized system validation (CSV): is any instrument qualified, calibrated, and any software validated? It is this layer of the model that this three-part series is focused on because it is fundamental to the success of data integrity in a regulated chromatography laboratory.
Regulatory Requirements and USP <1058>
The US GMP regulations for equipment are found in section 211.63 (6):
Equipment used …. shall be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance.
The equivalent EU GMP regulations are in Chapter 3 clause 3.34 (7):
Manufacturing equipment should be designed, located, and maintained to suit its intended purpose.
Both US and EU regulations require adequate design for intended use, adequate size, and suitable location. Intended use has been interpreted as documenting requirements, otherwise. how can you determine what the use will be and verify that it works?
The updated version of USP <1058> on Analytical Instrument Qualification (8) has the following statements:
The first activity is the generation of a user requirements specification (URS), which defines the laboratory’s particular needs and technical and operational requirements that are to be met.
The subsequent qualification activities necessary to establish fitness for purpose may be grouped into four phases: design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ).
Relationships Between the 4Qs Phases
The relationships between the various qualification phases can be seen in Figure 2. In this column we will look at two of these relationships:
- User Requirements Specification and Design Qualification
- Operational Qualification and User Requirements Specification.

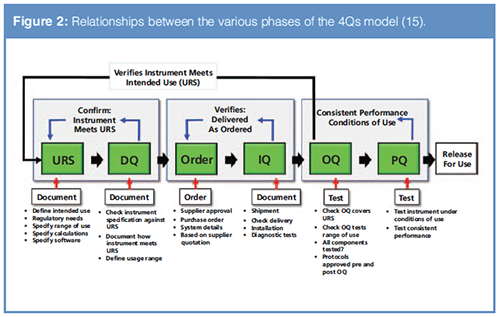
Understanding the URS and DQ Relationship
Writing the URS for an analytical instrument is a totally separate activity from the DQ phase. In separating the two activities, USP <1058> harmonizes with clauses 3.2 and 3.3 in EU GMP Annex 15 (9). DQ is an activity that confirms that the instrument proposed for purchasing meets the requirements in the URS. The requirements in the OQ phase of work are tested as discussed in the January QOQ column (1).
The problem now comes with a further statement in the new USP <1058> (2):
It is expected that DQ requirements will be minimal for commercial, off-the-shelf instruments.
Note these words well. Requirements will be minimal. Not zero, not virtual, not imaginary, not verbal but minimal. Minimal = written down, documented, recorded, and formal (approved).
Don’t Forget the Software Specification!
Although USP <1058> says that a URS for a chromatograph is expected to be minimal, what about any associated chromatography data system software used to control the instrument and also acquire, process, store, and report data? Minimal? We don’t think so. We must factor in data integrity requirements as well as the software functionality for the intended use of the instrument. This is not an exercise in minimalism (see reference 4). The problem is that all the regulations, for example, GMP, GLP, Part 11, were nearly all written before data integrity became a major issue, and regulators are now relying on guidance documents to define their requirements for the integrity of data. This year the EU GMP Annex 11 and Chapter 4, on computerized systems and documentation respectively, will be updated specifically to expand requirements and controls expected for ensuring data integrity into law and not rely on guidance.
Configuration of software is important for computerized laboratory systems because the intended use of the software including data integrity can only be tested when the application settings have been enabled and documented. For example, segregation of duties, enabling record protection, and turning the audit trail on. A major problem is that chromatography instruments and software can be ordered without involving the QA data integrity group whose function should be to see if an application is suitable for use in a regulated environment.
Instrument DQ and Supplier Assessment
Once the laboratory URS has been written, it is time to assess what is on the market and compare the supplier’s specification with what has been written. How the supplier has measured each of the parameters in their specification must be understood (2). The new way of performing DQ is the comparison of the laboratory URS with the supplier’s specification.
Although the focus is on the instrument, never forget that the second element in the equation is the instrument supplier, each one should be assessed. USP <1058> notes that part of DQ is “users should also determine the supplier’s capability to support installation, services, and training” (2).
The main topics to consider are the quality of the IQ and OQ documents, technical support, maintenance, and the quality of the training they provide to their engineers. It is essential that a supplier is thought of as an extension of the laboratory and an essential business or compliance partner and as such the thinking must change from “them” and “us” to “we”. However, this means that a more rigorous assessment and approval process is required if a supplier’s instrument and services are to be used effectively and not leave any compliance gaps.
Requesting an instrument supplier perform qualification services falls under EU GMP Chapter 7 on Outsourcing (10) and an agreement covering the services and the roles and responsibilities of both the laboratory and the supplier is required.
URS and OQ: Developing an OQ Protocol
One of the issues when designing an OQ protocol for a chromatograph is to know the parameters to qualify and the operational ranges and the uncertainty limits to specify. The laboratory URS for the instrument as required by USP <1058> (2) will define the parameters and operational ranges for each. A recent Focus on Quality column in Spectroscopy goes into more detail about writing a URS for an instrument (11). A question that arises is what should be the uncertainty associated with each parameter for the qualification?
Typically, a manufacturer’s specification for an analytical instrument is much tighter than the regulatory limits that need to be applied. When selecting an instrument, the DQ should document that the supplier’s specification satisfies the URS. Be careful as a manufacturer’s specification can create uncertainty over what limits need to be used during the qualification. As a broad generalization, the qualification limits selected should be the same as the regulatory requirements and it is a laboratory responsibility to ensure that this is the case. However, it can also depend on the quality and thought that has been applied to the URS and associated life cycle process that documents how the URS will be tested.
Some laboratories may tend to copy the supplier instrument specification or apply pharmacopoeial drug testing limits, such as injection precision limits that apply when performing a pharmaceutical analysis. An instrument will be tested to its manufacturing specification before it leaves the factory, but it is possible that the instrument will not be able to pass the manufacturing specification throughout its life time of use.
Some laboratories believe that it is a good practice to apply very tight limits to qualification testing. The rationale is that if the chromatograph passes very challenging test limits, there will be greater confidence in the instrument working correctly when it is used routinely. This is a grave mistake. Why test an instrument to a tighter specification than required by the regulations? There is a greater possibility that an instrument will fail the qualification test, delaying the return of the instrument to routine use and creating additional work reviewing the potential impact of the failure.
The relationship between the URS, DQ, OQ, PQ, and point of use testing such as system suitability testing (SST) needs to be very well understood because under the new USP <1058>, PQ testing now extends into routine use of the instrument (2).
Executing an OQ Protocol
If an instrument fails a qualification test, how “visible” is the failure in the qualification protocol and to the laboratory and how was the failure “managed”? The FDA has cited data integrity-related examples of poorly managed qualification failures where repeating failed tests have resulted in the instrument being tested into compliance (12).
During the qualification of HPLC system 10, four consecutive tests were performed until a passing result was achieved.
There can also be discrepancies between the protocol and the data that supports it as shown in an FDA warning letter:
Our inspection revealed discrepancies between the printed chromatograms and the operational qualification protocol for the High Performance Liquid Chromatography (HPLC) system, which is intended to demonstrate correct operation of the HPLC. These discrepancies included injection sequences and values to calculate relative standard deviation (RSD) (13).
If the qualification is being performed by a service provider and there is a qualification test failure, should all work on the instrument be stopped until the engineer can find a responsible manager to agree on the investigation and the next steps? Ideally, how to manage, investigate, and document test failures and record protocol deviations should have been discussed in detail and actions and responsibilities agreed. However, in practice, it is quite rare for quality agreements to be defined with this level of granularity and this represents a risk to both the service provider and the laboratory. A more pragmatic approach would be to support criteria under which qualification tests can be repeated, such as where there is an appropriately documented and verifiable reason for the test failure.
OQ Protocols: Flexible vs. Standard
One of the issues when selecting a suppler is assessing the quality of the qualification documentation. Remember from the overview of the changes in USP <1058> (1) that the OQ must be performed against the requirements in the laboratory URS: does the supplier’s OQ cover all requirements? Bob’s thinking has not changed in 20 years and here is a quotation:
The key to deciding if vendor material is useful to your laboratory is to assess the tests carried out against the design specification. If the vendor’s material matches all your key parameters, the vendor’s material should be suitable for your use. However, in our experience, this is rarely the case as laboratories often operate in ways that are not covered by the vendor’s generic documentation (14).
If the OQ protocol covers all parameters documented in the URS, then outsource the work to the supplier. However, if there are gaps then additional testing must be carried out to fill them, or carry a regulatory finding and hope an inspector does not find out.
Where flexible OQ protocols are used and are configured to match the URS, it is important that the laboratory understands the process the supplier follows to develop, test, and validate the protocols they support. Asking a service provider to perform qualification testing that uses protocol set points or limits that are outside of the life cycle process is effectively asking them to perform a qualification service that is outside of the range they have validated in the protocol development (always check that protocol development follows a validation life cycle process).
Wavelength Qualification Below 205 nm
There are currently no suitable reference materials that can be used to test the wavelength accuracy of a UV–vis HPLC detector below the 205-nm peak of caffeine. Therefore, if a laboratory has HPLC methods that operate below 205 nm, the accuracy of the detector cannot be directly measured below 205 nm. A new reference material is available for
UV–vis spectrophotometers using matched quartz cuvettes, but this is currently not suitable for HPLC detectors. Holographic gratings are typically used within HPLC detectors because of their greater resolving power. This means that it is extremely unlikely the detector would fail at 200 nm if other wavelengths are tested and pass because of the linear nature of the grating, but this would need to be documented and approved.
Change of Instrument Use
One of the consequences from the new version of <1058> is that the AIQ process is now a dynamic activity. Change the use or intended use of an instrument and it will have implications for the AIQ life cycle (for example, it may change the group classification; the URS, DQ, and OQ may need to be changed in order to ensure the use of the system is qualified). This means that change control processes associated with updating and approving qualification documents must be efficient or there is a risk of noncompliance.
Impact of AIQ on Data Integrity
From the data integrity model, AIQ and CSV are essential to ensure the correct measurement of instruments and operation of software. When you know that the instrument and software work as intended you can have the confidence that the analytical procedures developed and applied generate quality data where the integrity is assured.
Conclusions
In the first of a three-part series looking at new version USP <1058> and how it can help ensure data integrity, how to write adequate specifications for analytical instrument qualification and the section of a supplier has been discussed. OQ protocols need to test the ranges of each parameter in the laboratory URS. In the next article in this series, the execution of the protocol and how the supplier’s service engineer should be supervised will be discussed.
References
- R.D. McDowall, LCGC Europe31(1), 36–41 (2018).
- United States Pharmacopeia General Chapter <1058> “Analytical Instrument Qualification,” (United States Pharmacopeial Convention, Rockville, Maryland, USA).
- EudraGMDP Report 35705 for Pharmaceutics International LLC. 2016. Available from: http://eudragmdp.ema.europa.eu/inspections/gmpc/searchGMPNonCompliance.do.
- R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Second Edition, Royal Society of Chemistry, Cambridge, UK, 2017).
- R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, UK, In Press)
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Silver Spring, Maryland, USA, 2008).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 3 Premise and Equipment (European Commission, Brussels, Belgium, 2014).
- General Chapter <1058> “Analytical Instrument Qualification” in United States Pharmacopeia 41 (United States Pharmacopeial Rockville, Maryland, USA, 2018).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation (European Commission, Brussels, Belgium, 2015).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 7 Outsourced Activities (European Commission, Brussels, Belgium, 2013)
- R.D. McDowall, Spectroscopy33(4), 12–16 (2018).
- FDA 483 Observations, Bayer, Germany, 2016.
- FDA Warning Letter VUAB Pharma ss. 2015. Available from: https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm448433.htm.
- C. Burgess, D.G. Jones, and R.D. McDowall, Analyst123, 1879–1886 (1998).
- R.D. McDowall and P. Smith, How to Comply with the 2017 Version of USP <1058>, Agilent Publication 5991-9419EN, (In Press).
Paul Smith is a Global Strategic Compliance Specialist at Agilent Technologies. After initially specializing in spectroscopy and application of chemometrics to spectroscopic data, Paul developed his compliance expertise in a variety of quality and management roles within the 17 years he spent in the pharmaceutical industry. Paul worked as an independent consultant and university lecturer before moving into laboratory compliance consultancy roles.
“Questions of Quality” editor Bob McDowall is Director at R.D. McDowall Ltd., Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the editor-in-chief, Alasdair Matheson, at alasdair.matheson@ubm.com








What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






