Combining Orthogonal Comprehensive Two-Dimensional Liquid Chromatography Methods in Food Analysis
LCGC Europe
This review illustrates the benefits of LC?C in food analysis and discusses the combination of LC modes used in the two dimensions, and the nature of the samples analyzed.
Comprehensive two-dimensional liquid chromatography (LC×LC) offers much higher peak capacities than single dimension separations. The degree of orthogonality between both dimensions is a critical factor for obtaining higher values for peak capacity. This review illustrates the benefits of LC×LC in food analysis and discusses the combination of LC modes used in the two dimensions, and the nature of the samples analyzed.
One-dimensional (1D) chromatography is used widely in a variety of applications. However, this approach often does not provide adequate resolving power for the analysis of complex mixtures found in biological, food, environmental, and pharmaceutical sciences. One potential solution to overcome this limitation is to use multidimensional liquid chromatography (2D LC), where the two dimensions are based on different separation mechanisms (1–4).



(PHOTO CREDIT: T_KIMURA/GETTY IMAGES)
The distinction between multidimensional LC (MD LC) techniques into "heart-cutting" and "comprehensive LC", abbreviated as LC–LC and LC×LC, respectively, is generally agreed upon in the chromatography community (5). In both approaches, the columns are connected by a switching valve. The main difference between these two techniques is the amount of the primary column effluent that is transferred. In the former approach, only a few selected fractions of the effluent containing the analytes of interest are directed from the primary to the secondary column (dimension) (6,7); in LC×LC, the entire sample is subjected to separation in both dimensions (8).
Furthermore, 2D LC can be performed either "off-line" or "on-line", depending on how the first dimension (1D) effluent is transferred to the second column (2D). Off-line 2D LC is the most popular approach because it is easier and very simple to develop. Fractions of the 1D effluent are collected manually or using a fraction collector, and then are re-injected on the secondary column. However, the "off-line" approach can be time-consuming, difficult to automate, non-reproducible, and susceptible to sample loss, contamination, and artefact formation. The "on-line" approach is faster and more reproducible, but it requires an interface and is more difficult to operate (1–4). Further requirements of a comprehensive 2D separation are that any two components separated in the first dimension must remain separated in the second dimension and that elution profiles from both dimensions are preserved (9,10). A 2D LC separation is considered "orthogonal" if the two separation mechanisms are independent of each other, therefore providing complementary selectivities. The sample components are spread out through two different retention patterns and over a range as broad as possible for retention factors (11,12).
Successful orthogonal separations can be achieved when suitable mobile and stationary phases are selected, taking into account the physicochemical properties of the sample components including size and charge, hydrophobicity and polarity. In particular, LC techniques offer a wide variety of separation mechanisms, such as normal-phase, reversed-phase, size-exclusion (SEC), ion exchange (IEX),and affinity chromatography (AC), which are characterized by different selectivities. A related benefit is the great identification power, which results from the formation of 2D chemical class patterns. These unique capabilities have attracted many researchers, who have applied comprehensive LC methods to charecterize their samples. The first comprehensive two-dimensional liquid system was introduced by Erni and Frei (13) for the analysis of a complex plant extract with a SEC column in the 1D and a reversed phase column in the 2D. In the last two decades, several LC×LC methods have been developed and applied to bio-active molecules contained in food samples (14–59). Specific reviews in the field of LC×LC applied to bio-active samples have been published covering both theoretical and practical aspects (1–4,60–69). To begin with we will briefly focus on the general aspects that should be considered when developing an LC×LC method. We will then focus on selected food applications using LC×LC, according to the combination of LC modes used in the two dimensions and the nature of the samples analyzed.



Method Development and Instrumentation in LC×LC
The development and optimization of an LC×LC method requires the adjustment of many parameters to achieve successful separations and satisfactory results. An LC×LC system is composed of at least two pumps, two columns, an injector, an interface, and a detector. The interface hyphenates the two dimensions and in the most common set-up, small-volume fractions of the effluent from the 1D are transferred using a multi-port switching valve into the 2D. Before coupling, the sample characteristics and the parameters that affect the peak capacity should be considered to optimize the method.
For the 2D analyses, separation time should be fast enough to ensure both complete fraction elution before the subsequent transfer and adequate 1D sampling (10). This time is slightly enhanced if a regeneration step needs to be considered when running gradient programmes. After defining the 2D analysis time, the 1D analysis time can be optimized at a low flow rate to get three to four samplings for each peak (10). It is ideally achieved using a gradient programme that maintains constant peak widths during the run.
To increase the analysis speed in the 2D, several approaches have been proposed. Monolithic columns have been extensively investigated because of their short regeneration characteristics and high permeability to allow operation at high flow rates without a loss in resolution (14–18,23–26,30,33,34,70–74). Another way to speed-up the 2D analysis is to use conventional columns packed with stationary phases of reduced particle size, such as partially or superficially porous stationary phases (19,20,27,28,38,39,41–44,46,49) or sub-2-μm particle packed columns (50); however, such an approach is not compatible with conventional LC instrumentation (PMAX = 440 bar) and requires more sophisticated hardware capable of withstanding such high pressures (ultrahigh-pressure liquid chromatography [UHPLC]) (28,50). To speed up the 2D analysis, the use of elevated temperatures with ultra-fast gradients in the 2D have been exploited (3). In fact, such an approach allows the flow rate to be increased thanks to the reduced mobile phase viscosity; however, the major concern is the stability of the stationary phase and analytes.
Both dimensions can be operated under either isocratic or gradient conditions. The latter can be considered beneficial because it avoids "wrap-around phenomena" that result from incomplete elution of fractions in the 2D analysis from an unmatch with the sampling time; however, rapid changes of the mobile phase composition may have negative effects on the background signals of UV or mass spectrometry (MS) detectors.
Many configurations have been designed and used in different applications as an interface for the automatic transfer of the 1D effluent to the 2D. The most widely used interface involves the use of a 2-position/10-port switching valve (14–20,22–27,29–44,46,49,54–58); a 2-position/8-port switching valve (75–77) or two 2-position/6-port switching valves have been used as well (28,47,50,59). The loops are alternately emptied and filled with the 1D effluent prior to the 2D analysis in a "continuous" way.
However, "stop-flow" methods in the 1D LC×LC system have been applied as well, whenever the 2D separation could not keep up with 1D sampling frequency (29). Such an approach allows the use of a longer 2D column, leading to enhanced resolution and peak capacity. The empty storage loops can be replaced by loops packed with stationary phase. Under these conditions, the 1D mobile phase is preferably a weak solvent and the solutes are focused in the loop before being transferred to the 2D; fast desorption by a strong solvent is performed afterwards by the 2D mobile phase (30,33). In addition, two parallel 2D columns can be used in the absence of storage loops. In such a case, columns of the same batch and with the same length of tubing need to be used (31,32,40).
Many technical issues can arise from the development of an LC×LC system. Firstly, the transfer of the effluent from the 1D to the 2D must be performed in a fast and reliable way. Furthermore, the volumes of tubing, column connections, and the internal parts of the valve ports should be reduced to avoid extra-column band broadening. For such an issue, the 1D usually consists of micro- (1.0-mm i.d.) or narrow-bore (2.1-mm i.d.) columns, which provide flow rates compatible with conventional 4.6-mm i.d. columns in the 2D. Another important issue is the compatibility of the mobile phases used in the two dimensions.
The mobile phase eluting from the 1D column should preferably consist of a weak solvent constituent of the 2D mobile phase to create a satisfactory peak compression and achieve "peak focusing" (74). Such a requirement is a must, whatever combination of LC mobile phases is used, particularly if the solvents or solvent mixtures are not completely miscible.
To this end, the coupling of normal-phase LC and reversed-phase LC is highly beneficial in terms of orthogonality, but not for solvent immiscibilities because the 1D mobile phase is usually apolar and normal-phase solvents are mixed with the aqueous 2D mobile phases, which can deteriorate the separation and lead to signal interferences (14–17,20,23–28,35). To solve this issue, a vacuum evaporation interface has been investigated (73). In this approach, the use of elevated temperatures under vacuum enabled not only the collection of the fractions eluting from the 1D via the switching valve, but also assisted the evaporation of the mobile phase solvents. When fully compatible 2 D solvents are used in reversed-phase LC × reversed-phase LC separations, flow instability is usually observed when a fluid of low viscosity displaces and penetrates a high viscosity solvent (viscous fingering) (78,79). For example, when acetonitrile and methanol are used as the mobile phases in the two dimensions of a reversed-phase LC × reversed-phase LC separation, viscous fingering occurs when the less viscous acetonitrile penetrates into methanol (4). Another recent promising column combination in LC×LC uses hydrophilic interaction liquid chromatography (HILIC) and reversed-phase conditions in the 1D and 2D, respectively. In the last two separation mode combinations, the mobile phase used in the 1D has a higher elution strength than the one used in the 2D. For this reason, the use of micro-flow rates in the 1D is highly beneficial to reduce dilution and provide flow rates compatible with 2D injection volumes (22,53–58).
Commercial ready-to-use LC×LC systems are currently available from various manufacturers, making the methodology much easier to adopt than when only self-assembled systems were used.
As far as detection is concerned, all conventional LC detectors, such as photo-diode array (PDA), mass spectrometry (MS) and evaporative light scattering (ELS) detectors can be used in LC×LC. Usually, a single detector is installed after the 2D column (although an additional detector can be used to collect the 1D data) with the 1D separation monitored only during the optimization step.
The high speed of the 2D analysis requires a very fast detector acquisition rate to ensure adequate sampling, which is critical for quantification purposes, otherwise a loss in resolution may occur. MS can identify the co-eluting non-isobaric peaks when they are not resolved by chromatography, and can be considered a third dimension to the LC×LC system. Electrospray ionization (ESI) and atmospheric-pressure chemical ionization (APCI) are used more often for on-line analyses, while matrix-assisted laser desorption ionization (MALDI) is usually applied to off-line collected fractions. In all cases, the 2D effluent must allow ionization of all sample compounds.
Despite an ever-increasing number of LC×LC applications, quantification experiments have rarely been described (36,37,41,49). Such scanty quantitative applications in LC×LC separations could be a result of the unavailability of dedicated software, therefore demanding considerably longer time for data elaboration. It should also be noted that most developments have been performed for software applied to gas chromatography (GC)- based instrumentation rather than LC-based systems. One of the main reasons is the much more complicated background signal in 2D LC than those in 2D GC, at least for UV absorbance detection compared with flame ionization detection (FID) (80).
Comprehensive LC Techniques in Food Analysis
Several LC modes can be investigated in LC×LC to suit specific separation problems. Normal-phase LC × reversed-phase LC, reversed-phase LC × reversed-phase LC, and HILIC × reversed-phase LC systems have been developed and applied to the analysis of food samples. In normal-phase LC mode, two different separation methods were explored for carotenoid analysis: Adsorption with a polar inorganic packing material, generally silica (23,24), and partition, in which a polar organic phase is chemically bonded to the silica substrate, such as cyanopropyl (CN) (25–28) or diol groups (35). Among normal-phase LC × reversed-phase LC approaches, a partition mechanism can be considered for the combination of silver ion chromatography with non-aqueous reversed-phase chromatography (SIC × NA reversed-phase LC) employed for triacylglycerol (TAG) analysis (14–21). On the other hand, HILIC × reversed-phase LC and reversed-phase LC × reversed-phase LC has been used for the analysis of phospholipids (PLs) in milk samples (22) and polyphenols in various food-related products (29–34,36–39,41–46,48–50,53–59).
SIC × Reversed-Phase LC Applications for Triacylglycerol Analysis
Nearly all of the commercially important fats and oils of animal and plant origin consist almost exclusively of the simplest lipid class, triacylglycerols. These are characterized by a high number of individual species because of the numerous combinations of fatty acids (FAs) bonded to the glycerol backbone. In all applications investigated by SIC × NA reversed-phase LC (14–21), TAGs are separated in the 1D to increase double bond (DB) numbers, as well as to the position/configuration of the double bonds within each FA and in the 2D on the basis of increasing partition numbers: PN=CN-2DB, where "CN" relates to the total carbon number of the three FAs.
ELS and APCI–MS systems have also been used. Such a column combination has been investigated for the analysis of TAGs in donkey milk (15,17) and different vegetable oils, namely rice (14), soybean (16), linseed (16), corn (18), borage (19), and peanut (20,21). In all cases for the 1D separations, either a micro-bore (14–17,19–21) or a narrow-bore (18) strong cation exchange (SCX) column was subjected to a lab-argentation. For all 1D separations, n-hexane was used as the main LC solvent; an alternative was reported by Van der Klift et al. with a methanol-based solvent on a silver-coated ion exchanger, avoiding the use of n-hexane and enabling "peak focusing" at the head of the 2D column (18). In terms of detection, UV, ELS, and APCI–MS were compared with the aim of improving the accuracy and precision of TAG quantitation. More recently, a similar set-up was investigated for the analysis of a peanut oil sample. A total of 28 TAGs were identified on the basis of their retention behaviours and APCI–MS spectra as can be seen from the 2D plot in Figure 1 (20). The high resolution MS detector significantly improved the total peak capacity by adding an effective third dimension of separation. The same sample was later analyzed by Hu et al. by using an inverted set-up with NA reversed-phase LC in the 1D and SIC in the 2D. This approach led to the identification and quantification of 48 TAGs including regioisomers (21).


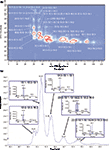
Figure 1: (a) LC Ã NA reversed-phase LC separation of peanut oil. (b) A raw chromatogram expansion relative to a single modulation in the same analysis. TAGs were denoted by the three fatty acids linked to the glycerol backbone and their positions did not represent the stereochemical positions. For details see (20).
Normal-Phase LC × Reversed-Phase LC Applications for Carotenoid Analysis
Carotenoids are among the most common pigments in nature and are usually characterized by a C40 tetraterpenoid structure, with a symmetrical skeleton. They are conventionally divided into two groups, namely hydrocarbon and oxygenated carotenoids, the latter present in a free form, or in a more stable, fatty-acid esterified form, depending on the presence or absence of oxygen in the structure. Because of their extreme instability, which leads to several molecular modifications, a much higher number of possible structures can arise and LC×LC separations represent an analytical challenge. Various LC×LC–PDA–APCI–MS systems have been investigated to elucidate the carotenoid composition of complex food samples, either on free carotenoids, attained after a saponification step (24), or on the native forms (25–28). In the first approach, a silica microbore column, operated under normal-phase LC conditions, was coupled to a C18 monolithic column, under reversed-phase LC conditions (24). In the second set-up, a cyano microbore column, operated under normal-phase LC conditions, was coupled to either a C18 monolithic (25,26) or partially porous column (27,28), under reversed-phase LC conditions. In the 1D, n-hexane/butylacetate/acetone 80:15:5 (v/v/v) and n-hexane were used, whereas 2-propanol and 20% water in acetonitrile (v/v) were used in the 2D. Under normal-phase LC conditions, free carotenoids are separated into groups of different polarity, from the non-polar carotenes up to the polar polyols. In the reversed-phase LC mode, carotenoids are eluted according to their increasing hydrophobicity and decreasing polarity. In all cases, the use of two detection systems, namely PDA and APCI–MS, allowed complementary information for positive carotenoid identification to be attained. This was despite the lack of commercial standard materials, and the fact that many carotenoids present very similar UV–vis or MS spectra. Additional information on carotenoids belonging to the same class can also be attained by considering specific peak positions in the 2D plots. In the most recent application on a Capsicum sample, the 2D of the LC×LC system was operated under UHPLC conditions by serially connecting two C18 columns of equivalent dimensions, reaching a 6-cm length of fused-core stationary phase (28). The performances of an LC×LC and two LC×UHPLC methods with different modulation times were evaluated in terms of peak capacity values and the results attained were corrected by considering both under-sampling and orthogonality effects. The best normal-phase LC × reversed-phase UHPLC approach turned out to be the one with a reduced modulation time, which resulted from better 1D sampling. A total of 33 compounds were achieved, belonging to 10 different classes. Detection was achieved by PDA and ion trap-time of flight (IT-TOF) MS (Figure 2).



Figure 2: Normal-phase LC Ã reversed-phase UHPLC analysis of free carotenoids and carotenoid esters in a red chili pepper extract with a modulation time of 1.50 min (PDA chromatogram extracted at 450 nm). The insets show the retention windows corresponding to (a) the di-ol-mono-keto-di-ester, (b) the di-ol-monoketo-mono-ester, and (c) the poly-oxygenated-free-xanthophyll classes, separated within a 1 min second-dimension gradient and modulation time. For details see (28).
HILIC × Reversed-Phase LC Applications for Phospholipid Analysis
Phospholipids are an important class of biomolecules that play a vital functional, structural, and metabolic role in the human body. They are classified into glycerophospholipids, which consist of a glycerol backbone esterified with two FAs at sn-1 and sn-2 positions, and the sn-3 position occupied by a phosphate group attached to a polar head of various structure; and sphingolipids, which are comprised of a sphingosine backbone, consisting of 18 carbon atoms, attached to the phosphate group.
Because of the polarity of PLs, normal-phase LC methods were widely used because retention is related to the polar head, that is, phosphatidylinositol (PI), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylcholine (PC), sphingomyelin (SM), and lysophosphatidylcholine (Lyso-PC). An alternative to normal-phase LC is represented by HILIC, which uses a hydrophilic stationary phase with an organic-dominant mobile phase. Its mechanism can be described as liquid–liquid partition chromatography, or a version of normal-phase LC, which can be performed with partially aqueous mobile phases. HILIC separates compounds by passing a hydrophobic or mostly organic mobile phase across a neutral hydrophilic stationary phase, causing solutes to elute in order of increasing hydrophilicity (81).
Each PL class is composed of a mixture containing many molecular species with different FAs; for this reason, reversed-phase LC techniques were exploited for PL separation, on the basis of FA chain length and degree of unsaturation.
On the basis of these observations, a single technique can only provide useful information on either the different PL classes, or the molecular species deriving from different PL classes. To fully exploit both of these features, a recent paper by Dugo et al. examined this (22). A novel LC×LC method with a silica HILIC column in the 1D and a C18 column in the 2D, based on stop-flow mode, in combination with ESI–MS detection was established to separate PLs in a Folch-extracted cow's milk sample. The enlargement of the HILIC × reversed-phase LC–ESI–MS contour plot, and the 2D raw data for the separation of PC molecular species is shown in Figure 3. The PC class was the richest in terms of molecular species, allowing a total of 16 PCs to be separated over two 15-min modulation cycles.

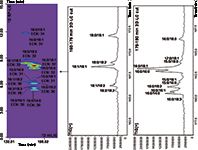
Figure 3: Enlargement of the HILIC Ã reversed-phase LCâESIâMS contour plot relative to the separation of the PC molecular species (cow milk), along with the corresponding 2D raw data. For details see (22).
HILIC × Reversed-Phase LC and Reversed-Phase LC × Reversed-Phase LC Applications for Polyphenol Analysis
Polyphenols are widely distributed in nature and have recently drawn considerable attention because of their possible health-promoting effects From a chemical viewpoint, they can be classified into different groups: phenolic acids, flavan-3-ols, flavanones, flavones, flavonoids, lignans, and so on (82). The complex structure of polyphenols, which occur particularly in fruit and plant extracts, requires powerful analytical methods to achieve rewarding results. To this end, HILIC × reversed-phase LC combinations have been investigated for polyphenol analysis in beverage and plant extracts (45,48,53–58), allowing compounds to be separated based on polarity and hydrophobicity, respectively.
De Villier and co-workers elucidated phenolics and procyanidins (PC) in apples and cocoa samples (45,57–58) and tea (48,53) extracts. In the first application an off-line LC×LC system was designed based on HILIC with a diol stationary phase and reversed-phase LC conditions in the 1D and 2D, respectively. Fluorescence, diode-array, and negative ESI–MS detection systems were used to unravel the complex composition of low-molecular-weight phenolic compounds.
The HILIC × reversed-phase LC system was characterized by a very high practical peak capacity (over 2000) because of the low degree of correlation (R2 <0.2) between the selected separation mechanisms. The method demonstrated its suitability for the analysis of various groups of phenolic compounds, including proanthocyanidins, phenolic acids, flavonols, and flavonol derivatives, all of which cannot be separated in a single analysis by conventional 1D LC methods. More recently, a comparison of practical peak capacities, analysis times and peak production rates was made for three different LC×LC configurations: on-line, off-line, and stop-flow (57).
In addition, the effects of 1D under-sampling, the degree of orthogonality between the two dimensions, and any additional band broadening were taken into account when using the stop-flow approach. In a separate contribution, the experimental verification of the findings of this study were reported for the analysis of cocoa procyanidins (58). The results showed that while optimization procedures based on theoretical considerations remain largely valid in practice, several important experimental considerations had to be taken into account to achieve maximum performance in all three modes of HILIC × reversed-phase LC. On the one hand, the on-line analysis provided an effective tool for the screening of procyanidin content within a reasonable analysis time, while the off-line and stop-flow HILIC × reversed-phase LC analyses were more suited for the detailed analysis of complex procyanidin fractions (Figure 4). In particular, stop-flow operation had a negligible effect on the 1D band broadening under the optimized experimental conditions used. Finally, off-line and stop-flow analyses provided much higher and similar resolving power at peak capacity production rates roughly half those obtained by the on-line system. However, the stop-flow system required the use of additional hardware and was experimentally more complicated, but it did offer the advantages of complete automation and minor risk of sample alteration. A similar set-up for PC analysis was also developed by Cifuentes and co-workers (55,56). The team investigated grape seed extracts using the optimized HILIC × reversed-phase LC separation, followed by PDA and tandem MS detection. This allowed the tentative identification of 43 flavan-3-ols, including monomers and procyanidin oligomers.



Figure 4: Fluorescence contour plots obtained for the HILIC Ã reversed-phase LC analysis of a cocoa extract using (a) off-line and (b) stop-flow configurations. Peak numbers indicate the degree of polymerization (DP) of PC isomers and letters differentiate isomers of the same DP. For details see (58).
The same set-up was also used for the phenolic profiling of different apple varieties in a timeframe of less than 50 min, allowing the tentative identification of approximately 65 compounds on each studied sample, including flavan-3-ol oligomers up to a DP = 8, dihydrochalcones, flavonols, and phenolic acids (56). This study opened up new possibilities for the application of procedures for other target and non-target metabolomics-related studies (56). Several polar stationary phases in the 1D, namely PEG, diol, amide, phenyl and sulphobetaine, were compared with gradients of decreasing concentrations of acetonitrile in buffered aqueous-organic mobile phases, and subsequently coupled on-line with short non-polar or weakly polar monolithic or porous shell columns in the 2D, with fast reversed-phase LC analyses (1–2 min) (54). For optimum performance of HILIC × reversed-phase systems, micro-bore or capillary columns and low flow rates were used in the 1D, while short (3-cm or 5-cm) core–shell columns with larger diameters were tested at high flow rates in the 2D. The 5-cm columns allowed the transfer of larger fraction volumes to the 2D without significant band broadening. For 1D a new monolithic sulphobetaine polymethacrylate capillary column was tested under HILIC conditions, providing better orthogonality and good efficiency, particularly for low molecular compounds; moreover, the 0.53 mm i.d. allowed fraction volumes to be lowered closer to the optimum for separations within 1 or 2 min fast gradients in the 2D.
Reversed-phase LC × reversed-phase LC has been the most widely applied method by far to the analysis of antioxidants and in particular polyphenols occurring in several types of food samples (29–44,46,47,49,50,52,59). However, in almost all reversed-phase LC × reversed-phase LC approaches investigated, "practical" peak capacity values were significantly lower than the theoretical values because of the coupling of partially correlated systems. An excellent way to overcome this issue was demonstrated by the use of careful gradient strategies in LC (46,83). Various types of 2D gradients in LC×LC were compared, namely "full in fraction", "segment in fraction", and "continuously shifting" gradients. In Figure 5 LC×LC separations of phenolic acids and flavones on a PEG column in the 1D and two types of partially porous fused-core C18 columns in the 2D using these gradients are illustrated. The effects of the gradient type on the bandwidths, theoretical peak capacity, separation time, and column pressure in the 2D were investigated.



Figure 5: 2D comprehensive LCÃC separation of phenolic acids and flavones on a PEG column in the first dimension and a partially porous fused-core C18 column in the second dimension. (a) Parallel gradients with the "full in fraction" (FIF) second-dimension gradient 1; (b) parallel gradients with the "segment in fraction" (SIF) second-dimension gradient 2; (c) parallel gradients with the "continuously shifting" (CS) second-dimension gradient 3. For details see (46).
A careful design of 2D gradients could be a valuable tool when partially correlated systems are employed in LC×LC separations.
Conclusions
Comprehensive LC has proven to be a viable tool for analysis of complex food samples thanks to the practical selection of separation systems with low selectivity correlations in the two dimensions.
The increasingly high interest in comprehensive LC in the last decade can be traced back to the commercial availability of ready-to-use LC×LC systems equipped with software for both method development and qualitative and quantitative data handling.
Further noteworthy improvements will be the exploitation of novel separation materials with very small particle size (sub-2-μm) and the development of analytical methods capable of reducing 2D analysis time for increased sample throughput and reducing 2D sensitivity loss with respect to the 1D.
Francesco Cacciola is an assistant professor of food chemistry at the University of Messina, Italy. His research interests include the characterization of complex food samples by conventional and innovative LC and MS techniques.
Marco Beccaria is a postdoc student at the University of Messina, Italy. His research field is related to the development of LC–MS and LC×LC–MS methods for lipid studies in food and biological samples.
Paola Donato is an assistant professor of analytical chemistry at the Campus Bio-Medico of Rome, Italy. The main objectives of her research activity involve the development of innovative LC techniques coupled to mass spectrometry (LC×LC–MS, LC×LC–MS–MS) for the analysis of highly complex mixtures.
Paola Dugo is a full professor of food chemistry at the University of Messina, Italy. Her research interests include the development of comprehensive liquid chromatography techniques, high throughput separation methods for the study of food components with potential biological activity.
Luigi Mondello is a full professor of analytical chemistry at the University of Messina, Italy. His research interests include chromatographic techniques, hyphenation to mass spectrometry, and multidimensional chromatographic approaches for the study of complex real-world samples.
References
(1) P. Dugo, F. Cacciola, T. Kumm, G. Dugo, and L. Mondello, J. Chromatogr. A 1184, 353–368 (2008).
(2) G. Guiochon, N. Marchetti, K. Mriziq, and R.A. Shalliker, J. Chromatogr. A 1189, 109–168 (2008).
(3) D.R. Stoll, X. Li, X. Wang, P.W. Carr, S.E.G. Porter, and S.C. Rutan, J. Chromatogr. A 168, 3–43 (2007).
(4) I. Francois, K Sandra, and P. Sandra, Anal Chim Acta. 641, 14–31 (2009).
(5) P. Schoenmakers, P. Marriot, and J. Beens, LCGC Europe 16(6), 335–339 (2003).
(6) J.M. Davis and J.C. Giddings, Anal. Chem. 55, 418–424 (1983).
(7) J.C. Giddings, J. Chromatogr. A 703, 3–15 (1995).
(8) L. Mondello, C. Lewis, and K.D. Bartle, Multidimensional Chromatography (John Wiley and Sons, Chichester, UK, 2001), pp. 109–129.
(9) J.C. Giddings, Anal. Chem. 56, 1258A–1270A (1984).
(10) Z. Liu, D.G. Patterson Jr, and M.L. Lee, Anal. Chem. 67, 3840–3485 (1995).
(11) P.J. Slonecker, X. Li, T.H. Ridgway, and J.G. Dorsay, Anal Chem. 68, 682–689 (1996).
(12) P. Jandera, LCGC Europe 20, 510–518 (2007).
(13) F. Erni and R.W. Frei, J. Chromatogr. 149, 561–569 (1978).
(14) L. Mondello, P.Q. Tranchida, V. Stanek, P. Jandera, G. Dugo, and P. Dugo, J. Chromatogr. A 1086, 91–98 (2005).
(15) P. Dugo, T. Kumm, M. Lo Presti, B. Chiofalo, E. Salimei, A. Fazio, A. Cotroneo, and L. Mondello, J. Sep. Sci. 28, 1023–1030 (2005).
(16) P. Dugo, T. Kumm, M.L. Crupi, A. Cotroneo, and L. Mondello, J. Chromatogr. A 1112, 269–275 (2006).
(17) P. Dugo, T. Kumm, B. Chiofalo, A. Cotroneo, and L. Mondello, J. Sep. Sci. 29, 1146–1154 (2006).
(18) E.J.C. van der Klift, G. Vivó-Troyols, F.W. Claassen, F.L. van Holthoon, and T.A. van Beek, J Chromatogr. A 1178, 43–55 (2008).
(19) L. Mondello, M. Beccaria, P. Donato, F. Cacciola, G. Dugo, and P. Dugo, J. Sep. Sci. 34, 688–692 (2011).
(20) Q. Yang, X. Shi, Q. Gu, S. Zhao, Y. Shan, and G. Xu, J. Chromatogr. B 895–896, 48–55 (2012).
(21) J. Hu, F. Wei, X.Y. Dong, X. Lv, M.L. Jiang, G.M. Li, and H. Chen, J. Sep. Sci. 36, 288–300 (2013).
(22) P. Dugo, N. Fawzy, F. Cichello, F. Cacciola, P. Donato, and L. Mondello, J. Chromatogr. A 1278, 46–53 (2013)
(23) P. Dugo, O. Favoino, R. Luppino, G. Dugo, and L. Mondello, Anal. Chem. 76, 2525–2530 (2004).
(24) P. Dugo, V. Skerikova, T. Kumm, A. Trozzi, P. Jandera, and L. Mondello, Anal. Chem. 78, 7743–7750 (2006).
(25) P. Dugo, M. Herrero, T. Kumm, D. Giuffrida, G. Dugo, and L. Mondello, J. Chromatogr. A 1189, 196–206 (2008).
(26) P. Dugo, M. Herrero, D. Giuffrida, T. Kumm, G. Dugo, and L. Mondello. J. Agric. Food Chem. 56, 3478–3485 (2008).
(27) P. Dugo, D. Giuffrida, M. Herrero, P. Donato, and L. Mondello. J. Sep. Sci. 32, 973–980 (2009).
(28) F. Cacciola, P. Donato, D. Giuffrida, G. Torre, P. Dugo, and L. Mondello. J. Chromatogr. A 1255, 244–251 (2012).
(29) E. Blahovà, P. Jandera, F. Cacciola, and L. Mondello, J. Sep. Sci. 29, 555–566 (2006).
(30) F. Cacciola, P. Jandera, E. Blahovà, and L. Mondello, J. Sep. Sci. 29, 2500–2513 (2006).
(31) F. Cacciola, P. Jandera, and L. Mondello, J. Sep. Sci. 30, 462–474 (2007).
(32) F. Cacciola, P. Jandera, and L. Mondello, Chromatographia 66, 661–667 (2007).
(33) F. Cacciola, P. Jandera, Z. Hajdu, P. Cesla, and L. Mondello. J. Chromatogr. A 1149, 73–87 (2007).
(34) M. Kivilompolo, V. Oburka, and T. Hyötyläinen, Anal. Bioanal. Chem. 391, 373–380 (2008).
(35) I. François, A. De Villers, and P. Sandra, J. Sep. Sci. 29, 492–498 (2006).
(36) M. Kivilompolo and T. Hyötyläinen, J. Chromatogr. A 1145, 155–164 (2007).
(37) J. Pòl, B. Hohnovà, and T. Hyötyläinen. J. Chromatogr. A 1150, 85–92 (2007).
(38) P. Dugo, F. Cacciola, M. Herrero, P. Donato, and L. Mondello, J. Sep. Sci. 31, 3297–3308 (2008).
(39) T. Hajek, V. Skerikova, P. Cesla, K. Vynuchalova, and P. Jandera, J. Sep. Sci. 31, 3309–3328 (2008).
(40) I. François, A. de Villiers, B. Tienpont, F. David, and P. Sandra, J. Chromatogr. A 1178, 33–42 (2008).
(41) P. Dugo, F. Cacciola, P. Donato, D. Airado-Rodriguez, M. Herrero, and L. Mondello, J. Chromatogr. A 1216, 7483–7487 (2009).
(42) P. Dugo, F. Cacciola, P. Donato, R. A. Jacques, E.B. Caramao, and L. Mondello, J. Chromatogr. A 1216, 7213–7221 (2009).
(43) P. Cesla, T. Hajek, and P. Jandera, J. Chromatogr. A 1216, 3443–3357 (2009).
(44) P. Jandera, T.Hajek, M.Stankova, K.Vynuchalová, and P.Cesla, J. Chromatogr. A 1216, 3443–3457 (2009).
(45) K.M. Kalili and A. Villiers, J. Chromatogr. A 1216, 6274–6284 (2009).
(46) P. Jandera, T. Hájek, and P. Cesla, J. Sep. Sci. 33, 1382–1397 (2010).
(47) M. Mnatsakanyan, P.G. Stevenson, D. Shock, X.A. Conlan, T.A. Goodie, K.N. Spencer, N.W. Barnett, P.S. Francis, and R.A. Shalliker, Talanta 82, 1349–1357 (2010).
(48) K.M. Kalili and A. Villiers, J. Sep. Sci. 33, 853–863 (2010).
(49) M. Russo, F. Cacciola, I. Bonaccorsi, P. Dugo, and L. Mondello, J. Sep. Sci. 34, 681–687 (2011).
(50) F. Cacciola, P. Delmonte, K. Jaworska, P. Dugo, L. Mondello, and J.I. Rader, J. Chromatogr. A 1218, 2012–2018 (2011).
(51) C.T. Scoparo, L.M. Souza, N. Dartora, G.L. Sassaki, P.A.J. Gorin, and M. Lacomini, J. Chromatogr. A 1222, 29–37 (2012).
(52) Q. Fu, Z. Guo, X. Zhang, Y. Liu, and X. Liang, J. Sep. Sci. 35, 1821–1827 (2012).
(53) T. Beelders, K.M. Kalili, E. Joubert, D. Beer, and A. Villiers, J. Sep. Sci. 35, 1808–1820 (2012).
(54) P. Jandera, T. Hajek, M. Stankova, Katerina Vynuchalova, and Petr Cesla, J. Chmatography A 1268, 91–101 (2012).
(55) L. Montero, M. Herrero, M. Prodanov, E. Ibanàñez, and A. Cifuentes, Anal. Bioanal. Chem. 405, 4627–4638 (2013).
(56) L. Montero, M. Herrero, E. Ibanez, and A. Cifuentes, J. Chromatogr. A 1313, 275–283 (2013).
(57) K. Kalili and A. De Villiers, J. Chromatogr. A 1289, 58–68 (2013).
(58) K. Kalili and A. De Villiers, J. Chromatogr. A 1289, 69–79 (2013).
(59) E.D. Larson, S.R. Groskreutz, D.C. Harmes, I.C. Gibbs-Hall, S.P. Trudo, R.C. Allen, S.C. Rutan, and D.R. Stoll, Anal. Bioanal. Chem. 405, 4639–4653 (2013).
(60) P.Q. Tranchida, P. Dugo, G. Dugo, and L. Mondello, J. Chromatogr. A 1054, 3–16 (2004).
(61) P.Q. Tranchida, P. Donato, P. Dugo, G. Dugo, and L. Mondello, Trends Anal. Chem. 26,191–205 (2007).
(62) P. Dugo, T. Kumm, F. Cacciola, G. Dugo, and L. Mondello, J. Liq. Chromatogr. Related Techn. 31, 1758–1807 (2008).
(63) H. Malerod, E. Lundanes, and T. Greibrokk, Anal. Methods 2, 110–122 (2010).
(64) M. Kivilompolo and J.T.Hyotylainen, LCGC Europe 24(5), 232–243 (2011).
(65) P. Donato, F. Cacciola, P.Q. Tranchida, P. Dugo, and L. Mondello, Mass. Spectrom. Rev. 31, 523–559 (2012).
(66) F. Cacciola, P. Donato, M. Beccaria, P. Dugo, and L. Mondello, LCGC Europe Supplement 25(5), 15–24 (2012).
(67) P. Jandera, J. Chromatogr. A 1255, 112–129 (2012).
(68) P.Q. Tranchida, P. Donato, F. Cacciola, M. Beccaria, P. Dugo, and L. Mondello, Trends Anal. Chem. 52, 86–205 (2013).
(69) P. Jandera, J. Chromatogr. A 1313, 37–53 (2013).
(70) A.M. van Nederkassel, A. Aerts, A. Dierick, D.L Massart, and Y. Vander Heyden, J. Pharm. Biomed. Anal. 32, 233–249 (2003).
(71) S. Eeltink G. Desmet, G. Vivo-Truyols, G.P. Rozing, P.J. Schoenmakers, and W.T. Kok, J. Chromatogr. A 1104, 256–262 (2006).
(72) O.P. Haefliger, Anal. Chem. 75, 371–378 (2003).
(73) H. Tian, J. Xu, Y. Xu, and Y. Guan, J. Chromatogr. A 1137, 42–48 (2006).
(74) N.E. Hoffman, S.-L. Pan, and A.M. Rustum, J. Chromatogr. A 465, 189–200 (1989).
(75) L. Hu, X. Chen, L. Kong, X. Su, M. Ye, and H. Zou, J. Chromatogr. A 1092, 191–198 (2005).
(76) X.Chen, L. Kong, X. Su, H. Fu, J. Ni, R. Zhao, and H. Zou, J. Chromatogr. A 1040, 169–178 (2004).
(77) X. Chen, Z. Jiang, Y. Zhu, and J. Tan, Chromatographia 65, 141–147 (2007).
(78) K.J. Mayfield et al. , J. Chromatogr. A 1080, 124–131 (2005).
(79) H.J. Catchpoole, R.A. Shalliker, G.R. Dennis, and G. Guiochon, J. Chromatogr. A 1117, 137–145 (2006).
(80) J.L. Adcock, M. Adams, B.S. Mitrevski, and P.J. Marriott, Anal. Chem. 81, 6797–6804 (2009).
(81) P. Donato, F. Cacciola, F. Cichello, M. Russo, P. Dugo, and L. Mondello, J. Chromatogr. A 1218, 6476–6482 (2011).
(82) A. Carrasco-Pancorbo, L. Cerretani, A. Bendini, A. Segura-Carretero, T. Gallina-Toschi, and A. Fernandez-Gutierrez, J. Sep. Sci. 28, 837–858 (2005).

















Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.



Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


