Column Re-equilibration Following Gradient Elution: How Long is Long Enough? Part I: Reversed-phase and HILIC Separations of Small Molecules
LCGC North America


Many users have questions about how long LC columns should be re-equilibrated following solvent gradient elution separations. Here we show satisfactory results with short re-equilibration periods, for both reversed-phase and HILIC separations.
Questions about how long LC columns should be re-equilibrated following solvent gradient elution separations are commonly raised and discussed by LC users. In this article, we discuss the details in light of recent research on this topic, and show that satisfactory results can be obtained with short re-equilibration periods on the order of two column volumes, even for HILIC separations.
Questions related to how long one must “re-equilibrate” a liquid chromatography (LC) column following a solvent gradient elution separation have been addressed in LCGC North America on several occasions, both in the past (1), and more recently (2). Answers to these questions have evolved over the past 20 years as a result of changes in instrument technology and characteristics, and deeper understanding of the factors that affect the behavior of columns under these conditions. Through many different conversations I’ve had with practicing chromatographers, it is clear to me that some of the findings of recent research in this area are not widely appreciated, so I think it is worth taking the time and space to continue addressing the topic, and with a pragmatic mindset.
For this month’s installment of “LC Troubleshooting,” I’ve decided to focus on questions about re-equilibration in reversed-phase LC as well as those specifically related to cases involving hydrophilic interaction chromatography (HILIC) separations of small molecules. I’ve asked Dr. Claudia Seidl to join me for this effort, because of her deep experience on this particular topic. Although there are still quite a few questions that are still unanswered about re-equilibration, and we await more research to address the gaps in the knowledge shared by the chromatography community, we hope that this installment facilitates the development of methods that are both effective and efficient.
Dwight Stoll
Introduction and Review
Core Concepts
When using LC in the solvent gradient elution mode, there must be a period of time, typically at the end of an analysis, when the mobile phase used in the first part of the solvent gradient is flowed through the LC column to prepare it for the next analysis. This period of time is commonly referred to as the re-equilibration time. The change in mobile phase composition with time is illustrated in Figure 1, where the re-equilibration time is denoted as tre-eq. Several important ideas are communicated by this figure. First, although the change in mobile phase composition (φi to φf ) is programmed (solid line) to begin at the start of the analysis, the onset of this change at the column inlet (dashed line) is delayed by the time, td, that it takes for solvent mixed by the pump to travel from the pump outlet to the column inlet. This delay affects not only the onset of the initial changes in composition from φi toward φf, but also from φf back to φi, as shown by the dashed line. Second, the shape of the dashed line on the right side of the figure shows that the return of the mobile phase composition from φf back to φi, as measured at the column inlet, does not follow the sudden step change that is programmed. Rather, there is a kind of exponential profile that results from the flushing of the instrument components situated between the outlet of the pumping system and the inlet of the column (for example, mixer, connecting tubing, and sampler loop). Recognizing that this exponential flush-out occurs is critical to understanding column re-equilibration in the context of solvent gradient elution. It is a matter of the physical behavior of the system that mobile phase with the composition φi does not begin flowing through the column again until long after the programmed end of the gradient. In Figure 1, we show that this occurs starting at the end of tflush. It is only at this point that the column can actually have a chance to equilibrate with the mobile phase used as the starting point in the solvent gradient (φi ). In practice we usually estimate that tflushis about two times the delay time (td ), or:


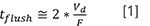
where Vd is the gradient delay volume, and F is the flow rate. Legacy pumping systems (for example, HP1100 or Alliance 2695) have gradient delay volumes on the order of 1 mL, whereas modern ultrahigh-pressure liquid chromatography (UHPLC) pumps have much smaller delay volumes on the order of 100 µL. If we consider a separation running at 0.5 mL/min, this means that tflush on a legacy system would be about 4 min, and, even on a modern UHPLC system, this time is not insignificant at 0.4 min. Again, the mobile phase delivered to the column inlet will not reach the initial composition used in the gradient (φi ) until after tflush, and so we cannot reasonably expect the column to equilibrate with this mobile phase until after this time has passed.

Figure 1: Solvent program used in gradient elution (solid line), and the mobile phase composition observed at the column inlet (dashed line). The change in composition is offset in time due to the delay time (td ) that results from the time it takes for a change in composition to travel from the mixing point to the column inlet. Eluent strength is for reversed-phase LC.
What Does It Mean For the Column to “Re-Equilibriate”?
Our first experiences studying this topic of column equilibration under gradient elution conditions came through work we did with Adam Schellinger and Professor Peter Carr at the University of Minnesota in the early 2000s (3–5). After recognizing the significance of the flush-out time, as discussed above, we realized pretty quickly that we needed to distinguish between two different levels or states of equilibration of the column:
- Full re-equilibration: This term refers to a state of the column where increasing the re-equilibration time (tre-eq ) further does not affect the retention times of analytes separated in a subsequent gradient elution separation. In other words, we can think of this state as one in which the column is truly equilibrated with the initial mobile phase used in the gradient (φi).
- Partial, repeatable re-equilibration: This term refers to a state of the column where it is not fully equilibrated with the initial mobile phase; however, highly repeatable separations can be obtained so long as the re-equilibration time used between gradient elution separations is highly precise.
The distinction between these two states of equilibration is important, because 1) getting to the state of full re-equilibration can take a long time (literally hours in some cases); and 2), in many situations, full re-equilibration of the column is not required to obtain a useful analytical result. In these cases, it is often far more important for the separation to be highly repeatable (for example, as measured by the precision of retention time) than it is for the column to be fully re-equilibrated, and if the analysis time can be reduced by using a re-equilibration time that is not unnecessarily long, then this can significantly improve throughput and reduce analysis cost.
We’ve Learned Some Things from Studying Re-equilibration of Reversed-Phase Columns in Detail
If we summarize a large amount of work carried out during our first deep look into the topic of re-equilibration of reversed-phase columns following solvent gradient elution, we end up with two principal conclusions:
- It is possible to obtain highly repeatable gradient elution separations with just one or two columns of equilibration with the initial mobile phase used in the gradient, after tflush has been reached.
- Progress toward full-equilibration can be affected by several factors, such as buffer concentration and pH, particularly for ionogenic analytes.
The first conclusion was quite striking to us at the time, because the prevailing thought at that time was that adequate re-equilibration of reversed-phase columns following solvent gradient elution required flushing the column with a minimum of 10 columns of the initial mobile phase used in the gradient program. If we consider the time savings between 10 and 2 column volumes of flushing, this can be substantial. For example, the dead volume of a typical 150 mm x 4.6 mm i.d. column is about 1.5 mL. If we use a flow rate of 2 mL/min, then the reduction of eight column volumes required for re-equilibration corresponds to 6 min of time that can be trimmed off a method that formerly required 10 column volumes of flushing for re-equilibration.
Our primary motivation for all of this work at the time was to better understand just how fast gradient elution could be done when used as a second dimension separation in two-dimensional liquid chromatography (2D-LC). Indeed, this improved understanding has led to the widespread use of conditions for 2D-LC that involve re-equilibration times for second-dimension separations that are just a few seconds in length, and correspond to a column volume or so of flushing with initial mobile phase (6,7).
What About HILIC Separations?
Realizing that highly repeatable results can be obtained with just one or two column volumes of flushing with initial mobile phase for reversed-phase columns, we can immediately think of several questions that are of practical interest. For example, in the April 2019 installment of “LC Troubleshooting,” we discussed the re-equilibration of reversed-phase columns with completely aqueous mobile phases following solvent gradient elution with acetonitrile. Here again, highly repeatable separations could be obtained following flushing with just one or two column volumes of aqueous mobile phase, whereas full re-equilibration required substantially more flushing (2).
Over the past few years, there has been considerable discussion about the rate of re-equilibration of HILIC columns. Very recently, David McCalley has used extensive experimental results to show that HILIC separations are not all that different from reversed-phase separations with respect to re-equilibration following solvent gradient elution (8,9). That is, highly repeatable HILIC separations can be obtained even after relatively short re-equilibration periods; however, full re-equilibration of HILIC phases can take a very long time (for example, tens of minutes or hours). The shortest re-equilibration time McCalley used in these studies was 4.3 min.
Most recently, we have explored the possibility that even shorter re-equilibration times could be used, while still yielding satisfactory HILIC separations. In our study, we examined the effect of re-equilibration time on analyte retention time and retention repeatability, covering an experimental space that included six different stationary phases, three mobile phase pH levels, different buffer concentrations, and both neutral and ionogenic analytes (10). In some cases we used re-equilibration times as short as 3 s. The overall conclusion from all of this work was that, consistent with McCalley’s observations, highly repeatable HILIC separations could be obtained with very short re-equilibration times. In our case, however, such highly repeatable separations were observed even at re-equilibration times as short as 3 s, provided that the re-equilibration time exceeded tflush under the conditions of the experiment.
Chromatograms Tell the Story
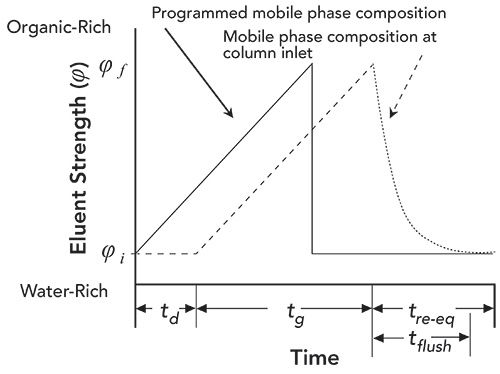
Figure 2 shows a series of chromatograms from our recent work on re-equilibration of HILIC columns. It is critically important to note here that these chromatograms were obtained using an unconventional instrument configuration, set up specifically for the purpose of this study, with two special characteristics: 1) we were able to precisely control the re-equilibration time between gradient separations (that is, to about 10 ms); and 2), the gradient delay volume was small (about 70 µL), but the maximum flow rate was still high for analytical columns (5 mL/min). Readers interested in these details are referred to the publication describing these results (10). The chromatograms shown in Figure 2 were obtained with re-equilibration times ranging from 0.3 to 15 min. Looking at these chromatograms, we see that there is very little shifting of retention for these analytes under these conditions as the re-equilibration time is changed, particularly over the range 2.5 < tre- eq < 15 min. The most significant change occurs for diphenhydramine (peak 2) as the re-equilibration time is reduced from 0.63 to 0.30 min. We see that the retention time of diphenhydramine increases slightly at the shorter re-equilibration time, resulting in a slight loss in the resolution of diphenhydramine and benzoic acid. Obviously this kind of dependence of resolution on re-equilibration time is important to know about. If discovered in the course of method development, one could avoid this sensitivity of the method by moving to a re-equilibration time of 1.3 min.

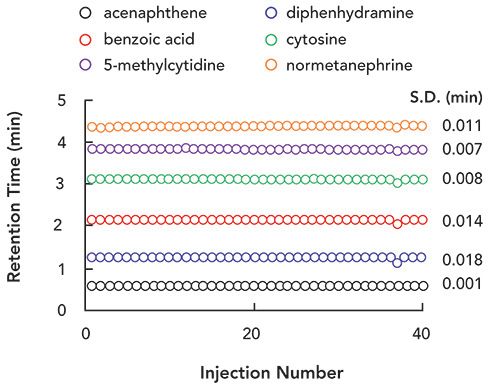
Figure 2: Effect of re-equilibration time (as 0.30, 0.63, 1.3, 5.0, 10, and 15 min) on retention time for small hydrophilic analytes separated by gradient elution on a HILIC column. For the 15-min. re-equilibration time, three replicate chromatograms are overlaid to provide a sense of the precision of retention with a given re-equilibration time. Chromatographic conditions: Column, AdvanceBio Glycan Mapping (50 x 2.1-mm i.d., 2.7-µm); Gradient, 95% to 80% B from 0 to 1.5 min. where mobile phase A is 100 mM ammonium acetate, adjusted to 6.0 with acetic acid, and B is acetonitrile; Temperature, 35 °C; Flow rate, 1.2 mL/min. Analytes: 1) acenaphthene, 2) diphenhydramine, 3) benzoic acid, 4) cytosine, 5) 5-methylcytidine, 6) normetanephrine.
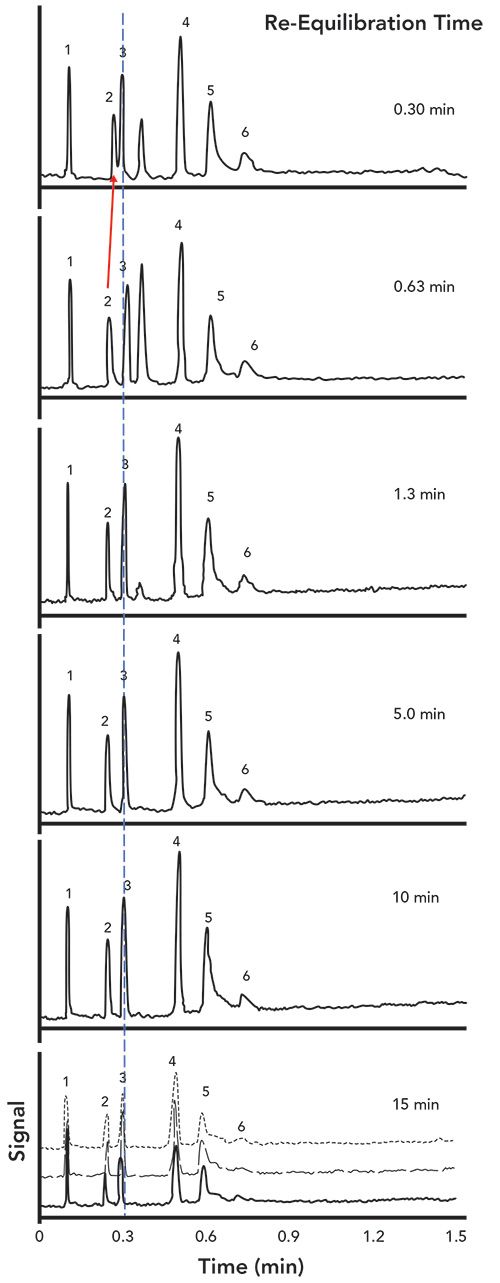
A more quantitative view of the same data is shown in Figure 3. Here, we have plotted the difference between the retention of a given analyte at a given re-equilibration time and the average retention of that analyte over all re-equilibration times. In other words, this view of the data shows us which analytes increase in retention as re-equilibration time is decreased, and which ones decrease. We see that, while the retention of most of the analytes is only weakly dependent on the re-equilibration time, the diphenhydramine retention increases significantly at the short re-equilibration time. The effect on the retention of methylcytidine and benzoic acid is measurable, but, in this case, not enough to affect the resolution of these analytes from their neighbors.

Figure 3: Quantitative view of the dependence of retention time on re-equilibration time for the same separations shown in Figure 2.
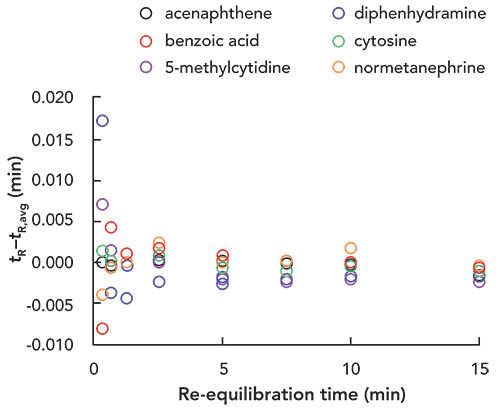
As stated above, most of our work on this re-equilibration topic has been done with instrumentation configured in an unconventional way that enables very precise control of the time between sample injections and gradient elution cycles. These results are immediately relevant to 2D-LC because second dimension separations are executed with a high degree of control over the time between separations. With conventional LC instrumentation, however, the user has less control over this time, due to data processing and other instrument initialization steps that occur between analyses, and so it is a fair question to ask how this variability in time between injections might affect the repeatability of retention for a HILIC method. To evaluate this, we set up a conventional LC instrument to do a separation similar to that shown in Figure 2, but with a longer column and slightly longer analysis time to emulate a more typical analysis. We then carried out 40 consecutive HILIC separations of the same six-component mixture shown in Figures 2 and 3 (gradient elution conditions are given in the caption to Figure 4) where the programmed re-equilibration time was set to just 30 s. The time between analyses was about 435 s, so the total re-equilibration time was about 75 s, which corresponds to about two column volumes under these conditions. The resulting retention times for these analyses are shown in Figure 4, along with the standard deviation of retention time for each analyte across the 40 separations. In this case, the time between injections was 436 ± 1 s for all 40 separations, which is pretty consistent, and does not tell us what the effect of more variation in the time between cycles might be (something like 5 or 10 s). Nevertheless, this result does show very convincingly that highly repeatable gradient elution HILIC separations can be obtained using just two column volumes of re-equilibration on a conventional instrument, provided that the re-equilibration time is longer than tflush.

Figure 4: Repeatability of retention time for a six-component mixture separated by HILIC using gradient elution. Chromatographic conditions: Instrument, Agilent 1290 UHPLC system with binary pump; Column, AdvanceBio Glycan Mapping (150 mm x 2.1 mm i.d., 2.7-µm); Gradient, 95-85-95-95 %B from 0-6.0-6.01-6.5 min. where mobile phase A is 100 mM ammonium acetate, adjusted to 6.0 with acetic acid, and B is acetonitrile; Temperature, 35 °C; Flow rate, 0.6 mL/min.
Conclusion
In this article, we have explained how high quality chromatographic results can be obtained for gradient elution separations even when the column is not truly equilibrated with the initial mobile phase used in the gradient. We have discussed how recent research has shown that this is possible even for HILIC separations, and with conventional LC instrumentation. There is no question that there will be situations and conditions where the use of very short re-equilibration times does not produce satisfactory results, and thus we suggest that during method development the re-equilibration time should be systematically varied to understand the dependence of retention and resolution on this variable. As John Dolan has wisely pointed out in previous discussions of this topic, a little too much re-equilibration is better than not enough (1). Nevertheless, significantly reducing re-equilibration times that would otherwise be unnecessarily long is a potential means of increasing sample throughput and decreasing analysis cost.
References
- J. Dolan, LCGC North Am. 21(10), 968–972 (2003).
- D.R. Stoll, LCGC North Am. 37(4), 244–249 (2019).
- A.P. Schellinger, D.R. Stoll, and P.W. Carr, J. Chromatogr. A 1192, 41–53 (2008). doi:10.1016/j.chroma.2008.01.062.
- A.P. Schellinger, D.R. Stoll, and P.W. Carr, J. Chromatogr. A 1192, 54–61 (2008). doi:10.1016/j.chroma.2008.02.049.
- A. Schellinger, D. Stoll, and P. Carr, J. Chromatogr. A 1064 143–156 (2005). doi:10.1016/j.chroma.2004.12.017.
- D.R. Stoll, and P.W. Carr, Anal. Chem. 89, 519–531 (2017). doi:10.1021/acs.analchem.6b03506.
- B.W.J. Pirok, D. Stoll R., and P.J. Schoenmakers, Anal. Chem. 91, 240–263 (2019). doi:10.1021/acs.analchem.8b04841.
- J.C. Heaton, N.W. Smith, D.V. McCalley, Anal. Chim. Acta 1045, 141–151 (2019). doi:10.1016/j.aca.2018.08.051.
- D.V. McCalley, J. Chromatogr. A 1554, 61–70 (2018). doi:10.1016/j.chroma.2018.04.016.
- C. Seidl, D.S. Bell, and D.R. Stoll, J. Chromatogr. A In Press (2019). doi:10.1016/j.chroma.2019.460484.


Dwight R. Stoll is the editor of “LC Troubleshooting.” Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 60 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com


Claudia Seidl is a postdoctoral associate in the Laboratory Medicine and Pathology Department of the University of Minnesota, in Minneapolis, Minnesota.


ct2019_MktPro_piechart_web.jpg")














Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

