The Chromatographic Society Meeting Report: Supercritical and Ultrahigh-Pressure Chromatography: Part 1
The three-day Chromatographic Society meeting was held on Monday 15th until Wednesday 17th May 2017 and was hosted by Pfizer at Discovery Park in Sandwich, Kent, UK. This comprehensive symposium featured oral presentations from leading practitioners of supercritical fluid chromatography (SFC) and ultrahigh-pressure liquid chromatography (UHPLC) from across Europe. Some of the latest innovations and applications were described and the lectures were augmented and supported by a comprehensive exhibition of instrumentation and consumables. The attendees gained insight into practical application of these techniques across a variety of industries, particularly the pharmaceutical industry. The comprehensive programme had a significant focus on SFC for chiral and preparative-scale analysis.


Photo Credit: plule_r/Shutterstock.com

Paul Ferguson, The Chromatographic Society
The three-day Chromatographic Society meeting was held on Monday 15th until Wednesday 17th May 2017 and was hosted by Pfizer at Discovery Park in Sandwich, Kent, UK. This comprehensive symposium featured oral presentations from leading practitioners of supercritical fluid chromatography (SFC) and ultrahigh-pressure liquid chromatography (UHPLC) from across Europe. Some of the latest innovations and applications were described and the lectures were augmented and supported by a comprehensive exhibition of instrumentation and consumables. The attendees gained insight into practical application of these techniques across a variety of industries, particularly the pharmaceutical industry. The comprehensive programme had a significant focus on SFC for chiral and preparative-scale analysis.
Around 100 scientists from industry, academia, and vendor organizations convened at Discovery Park in Sandwich, Kent, UK, from the 15th to 17th May 2017 to gain a better understanding of the latest applications of supercritical fluid chromatography (SFC) and ultrahigh-pressure liquid chromatography (UHPLC), review the state-of-the-art in these key separation approaches, and discuss separation science with peers through a very focused programme. The meeting was the Society’s Spring Symposium and also included its annual general meeting. As ever, The Chromatographic Society was indebted to the sponsors of the organization and the diverse range of vendors exhibiting at the meeting (see Table 1). This review is a concise account that does not cover all the presentations from the symposium, but it is hoped it will provide the reader with a flavour of the discussions from the meeting and the typical content of a Society meeting.


Some important trends in modern SFC and LC were found to be:
- SFC is becoming increasingly prevalent in industry for a variety of analytical applications because of the robust hardware, vendor qualification, and post-installation support allowing its application for GMP analysis.
- In the pharmaceutical discovery environment, the use of SFC for chiral and preparative separations is becoming so successful that LC approaches are becoming redundant. Success rates for chiral separations by SFC are routinely >95%.
- The impact of additives in SFC is becoming increasingly understood-particularly in chiral SFC.
- SFC is being utilized for analysis of complex or novel samples such as bile samples, vegetable matter, cosmetic allergens, or peptides.
- SFC–MS is extremely useful for synthetic chemistry reaction monitoring and can be incorporated into flow chemistry applications.
- For reversed-phase LC and HILIC separations, recent developments in silico retention prediction based on molecular structure is becoming more accurate.
- HILIC is moving to a position where the retention mechanisms are much better understood and effective screening protocols are commercially available from multiple vendors.
Day One
The symposium started on Monday afternoon with a blend of tutorials, demonstrations, and lectures. The conference programme is shown in Table 2. Tony Taylor of Crawford Scientific and ChromAcademy opened proceedings discussing the role of e-learning as a development tool for separation scientists. A team member from JayTee Biosciences then provided a laboratory-based session on high performance liquid chromatography (HPLC) and UHPLC best practice and instrument troubleshooting, which was well received by the attending delegates.

The final part of the day included two lectures from colleagues at Novartis. Both lectures were presented by Alexander Marziale. The first presentation was in the absence of John Reilly, who was unable to present by teleconference from their site in Boston, USA. The first talk was on “Open Access Purification to Support Discovery Chemistry” and focused on their comprehensive set of tools available for discovery chemistry support for over 200 chemists in Boston. The suite of services they offer covers purification, chromatography, mass balance, and physicochemical property determination. Mass balance service includes water determination and inorganics by ion chromatography (IC) (residual inorganics or salt stoichiometry determination). The physicochemical property determination tests include phospholipid, human serum albumin, and immobilized artificial membrane (IAM) partitioning assay. Alex highlighted how they have developed efficient open access LC–mass spectrometry (MS) and SFC–MS purification platforms aligned with analytical-scale high and low pH screening methods (for HPLC) and neutral and high pH methods for SFC screening. He noted that SFC was primarily used for polar molecules, fatty acids, and macrocyclic peptide compounds. In addition, open access chiral preparative SFC–MS was an extremely popular and important approach to supporting the chemists. In 2016, 120 chiral analyses were undertaken and 77 of these were performed through this approach.
Another avenue Novartis is pursuing is offering chemists with flash preparativeâscale services. For example, they have an approach using axial compression columns and 20-micron particles for chiral separations operating under normal phase conditions, which again allows more time for the nine analysts to support technical challenges of greater complexity.
Alex then moved on to present his work on “New Challenges for SFC in Drug Development”, which linked closely with the first presentation. He discussed how increasing comprehension of biological systems is providing exciting novel target classes, including protein–protein and protein–nucleic acid interactions, which are often “undruggable” by classic small molecule approaches. Novartis has focused interest on subjecting these novel compound classes to both analytical and preparative SFC and also uses analytical SFC to assess the polarity and cell-membrane permeability of these new modalities (such as cyclic peptides). Interestingly, this approach also enabled identification of intramolecular hydrogen bonds.
Day Two
The second day was opened by Roman Szucs (Senior Research Fellow and Pfizer meeting coâorganizer) and Brian Henry (VP, Drug Product Design, Pfizer) who discussed the history of Pfizer in Sandwich and how Discovery Park has transformed since Pfizer’s downsizing at the site in 2011. The site now has over 170 companies registered at the Sandwich-based science park with nearly 3000 personnel there-with the largest occupants being Pfizer and Mylan.
The first session of the day was provided by Tony Edge of Agilent Technologies. His presentation was titled “Supercritical Fluid Chromatography-No Regrets!” and was originally to be delivered by Keith Bartle-who unfortunately could not be at the event due to ill health. Tony discussed Keith’s long history in the field of separation science and some of his pivotal work in the fields of gas chromatography (GC) and SFC. He also acknowledged Keith’s role in the development of many famous chromatographers such as Ally Lewis (University of York), Paulo Dugo and Luigi Mondello (both University of Messina), and Tony Edge (Agilent/University of Liverpool) himself!
In an overview of Keith’s accomplishments, he noted his initial work on the supercritical fluid extraction (SFE) of oils from yew tree leaves-modelled using a “hot ball model”-noting that the process was diffusion limited. After discussions with James Lovelock (a past President of The Chromatographic Society) in 1958, Keith moved into the field of SFC. He also presented the separation of surfactants and hops using SFC with a density gradient, 25-µm capillary columns, and flame ionization detection (FID).
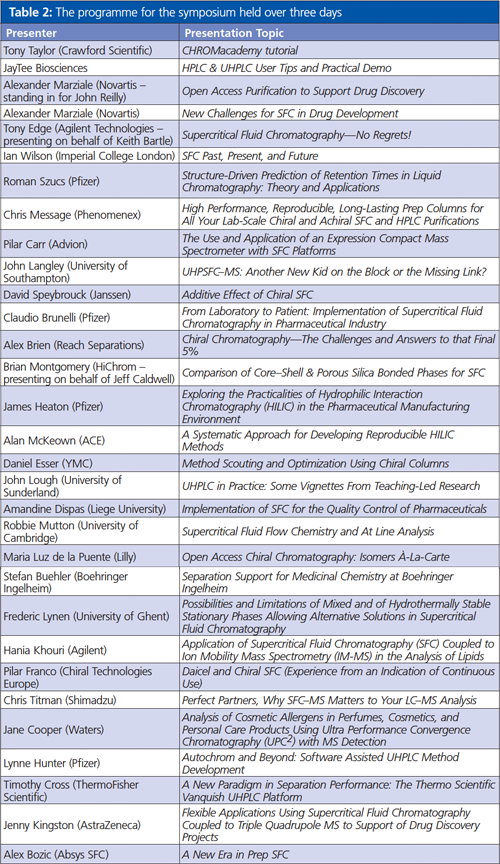
Keith also compared the separation of propylene glycol using SFC and size-exclusion chromatography (SEC). SFC offered a much more efficient separation albeit it in a much longer timeframe (see Figure 1). Keith also investigated the utility of other gases such as xenon for SFC for the separation of polymer oligomers using 50-µm capillary columns and Fourier-transform infrared spectrometry (FTâIR) detection, and the separation of diesel fuel with a helium mobile phase (10-m long column!) and FID detection.

Keith was always keen to unify separation theory across GC, HPLC, and SFC, and accordingly developed instrumentation to enable all three techniques to be used on the same single platform with propylene glycol cooling of the carbon dioxide pump heads. He illustrated this by coupling GC with SFC for the separation of volatile pharmaceutical compounds with FID. Keith’s research then moved onto packed column SFC-illustrating this through the separation of polyethylene glycol on hexyl-bonded silica with carbon dioxide pressure programming and FID detection. Moving back to HPLC, Tony discussed Keith’s work on enhanced fluidity chromatography. This was the use of HPLC mobile phases at increased temperatures and infused with carbon dioxide. He illustrated this through the separation of polyaromatic hydrocarbons and taxicins from yew tree extracts-this essentially being SFC with methanol as a solvent modifier.
The final areas Tony touched on was Keith’s work in preparative SFC and development of chiral molecularly imprinted phases (with Richard Ansell of Leeds University). Reiterating the areas he discussed (GC, SFC, subcritical fluid chromatography [SubCFC], enhanced fluidity chromatography, elevated temperature liquid chromatography [ETLC], liquid chromatography, and ultraHPLC), he asked if “Perhaps Keith and the other academics of that era had it right 30 years ago? And they have told us the future.”
The next speaker in this session was Ian Wilson (Imperial College London) who discussed “SFC, Past, Present, and Future” and his group’s work at the Metabalome Centre at the university. He started his presentation reflecting on the “second coming of SFC in the 1980s” and his work at ICI using this technique, which often consisted of homemade equipment (his being based on a GC column oven and LC pump coupled to an FID detector). He illustrated the success of this for the analysis of weak pharmaceutical acids (naproxen and ibuprofen) and the separation of weak bases (beta blockers) using amino and cyanopropyl columns with trimethylamine as a mobile phase additive (and surprisingly acetonitrile as organic solvent). He also showed examples of preparative SFC and the use of this technique for pharmaceutical radiolabel studies (with a radioactivity detector). He summarized this aspect of his research by stating that SFC worked well with diol, cyano, and aminopropyl columns in isocratic and gradient modes; the use of multiple detection approaches was possible; the technique was robust and cheap; and chiral separations looked a very promising application area.
He stated that although SFC was a very good separation technique it went out of vogue, and was replaced by capillary electrophoresis (CE) and capillary electrochromatography (CEC), but the lack of commercial availability of SFC instrumentation was also detrimental. However, as time progressed, preparative SFC became very popular, CE did not meet all expectations for small molecule analysis, and more companies entered the SFC market.
Ian went on to describe his group’s use of SFC for the profiling of dog bile and rat bile (1), which was found to be a useful approach for the separation of isobaric compounds such as tauroursodeoxycholic and taurodeoxycholic bile acids. These separations were found to be highly repeatable with very good retention time precision allowing metabolic phenotype profiling through principal component analysis (PCA). The principal test for Ian’s group was if SFC could be applied to the analysis of highly polar metabolites. Following analysis of 60 analytes under multiple different conditions to test this hypothesis, the columns chosen were organo-silane hybrid diol and 2-PIC (picolylamine) phases with methanol and isobutylamine as a mobile phase additive (which is MS compatible). This is being applied routinely in his laboratory for the analysis of complex sample types such as rat urine with good success.
In a late change to the programme, Roman Szucs (Pfizer) spoke on “Structure-Driven Prediction of Retention Times in Liquid Chromatography: Theory and Applications”. Roman introduced his presentation outlining the types of LC-based generic methods utilized by Pfizer for method development but highlighted that this was only one approach to generate a potential separative solution to an analytical challenge.
Roman described Pfizer’s efforts to predict chromatographic retention of analytes based on molecular structure under generic method conditions. Retention modelling approaches include the use of physicochemical properties, solvation parameters (“Abraham descriptors”), or molecular descriptors (such as those calculated through Dragon software: https://chm.kode-solutions.net/products_dragon.php). Roman’s group have found the first two approaches are useful if analytes have only one functional group.
The process used for calculating retention involves matching analyte molecular descriptors for known analytes (calculated through Dragon) with analyte retention times under the generic method conditions. Relevant descriptors are then selected using an algorithm (Tanimoto similarity index) and structure retention relationships determined and assessed using, for example, a partial least squares analysis of measured versus predicted retention times. The best fit mathematical relationship is then used to predict the retention of additional compounds under the same chromatographic conditions. Retention time errors were large (up to ±12.5%), but the approaches have been refined using smaller more structurally relevant data sets so that errors are routinely <4%. He stated that volume surface descriptor was a very important parameter for predicting retention.
Roman moved on to describe how this approach has been extended to hydrophilic interaction liquid chromatography (HILIC) retention modelling (2) and IC (3).
The next talk of the morning was given by Chris Message (Phenomenex) who discussed “High Performance, Reproducible, LongâLasting Lab-Prep Columns for HPLC and SFC”.
The final talk of the morning was provided by Pilar Carr (Advion) who discussed “The Use and Application of a Compact Mass Spectrometer with SFC Platforms“.
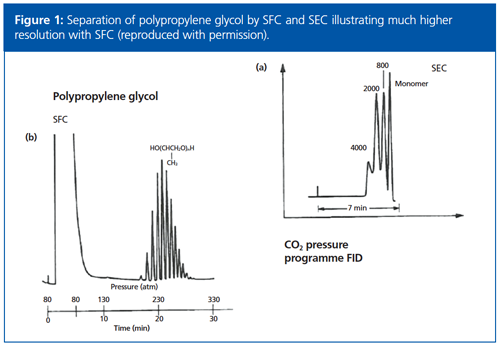
The Tuesday afternoon session was opened with an excellent presentation by John Langley (University of Southampton-see Figure 2) outlining the versatility of SFC–MS in his presentation “UHPSFC-MS: Another New Kid on the Block or the Missing Link?”. John opened his presentation by discussing the number of organic chemists, and the areas of research and instrumentation available to support them in the chemistry department at the university. The instrumentation includes open-access GC–MS and UHPLC–MS, but more recently SFC–MS has been introduced. He noted that while LC–MS is still the analytical workhorse in his department, SFC is being applied more for:
(i) When analytes do not exhibit retention by reversed-phase UHPLC–MS;
(ii) Early (and late) eluting compounds by reversed-phase UHPLC–MS;
(iii) Where there is sample incompatibility with reversed phase solvents;
(iv) To replace normal-phase LC literature methods;
(v) As an alternative to HILIC literature methods;
(vi) Separations requiring alternative selectivity.
John described some of the “interesting” types of samples they currently analyze using UHPLC–MS, such as microbial lipid extracts, those they analyze by GC–MS, such as first and second generation biofuels, and how these have been successfully moved to SFC–MS (4). He also described the use of SFC–MS for other impurity profiling activities, including the analysis of nucleotides, phosphoramidite building blocks for oligonucleotide synthesis, purine bases, sugars (see Figure 2), molecular wires (molecular chains that conduct electric current and are proposed building blocks for molecular electronic devices), and porphyrins amongst numerous other examples.

John concluded his presentation noting that SFC–MS is as easy to use as reversedâphase UHPLC–MS (maybe even easier) and it plays a crucial role in research at the university. He noted that while it is unlikely to replace other mainstream separation approaches, it is a complementary technique to reversedâphase UHPLC–MS and GC–MS and extends chromatography solutions to different chemistries and application areas.
David Spreybrouck from Janssen then spoke on “Additive Effects of Chiral SFC”. David jumped straight into his presentation by presenting their findings on the addition of acidic modifiers (0.3% [v/v] formic acid and TFA) to carbon dioxide–methanol mobile phases in the analysis of acidic compounds. They found that 96% of acidic compounds did not require acidic additives to elute them from amylose- and cellulose-based chiral stationary phases. Indeed, when additives were used, retention was observed to be lower than without. This was attributed to the acidic nature of the carbon dioxide mobile phase in the presence of water. He then discussed the addition of a basic additive to the mobile phase (0.3% [v/v] isopropylamine) for the analysis of acidic analytes. He found that for 90% of the compounds analyzed retention was lower than without an additive, but roughly a third of the compounds exhibited improved enantioselectivity with isopropylamine in the mobile phase. He also presented data showing that moving from 0 to 10% (v/v) isopropylamine (a huge amount of additive not typically used) in the mobile phase, elution order could be reversed for certain compounds.
He then moved on to discuss the impact of using additives with basic analytes in SFC. He reiterated the need for the use of additives for basic analytes or poor peak shapes will usually be observed. He presented data on the analysis of primary, secondary, and tertiary amines with up to 10% (v/v) isopropylamine in the mobile phase. For all amine types, retention was largely the same up to 10% additive, but again could have a pronounced effect on enantioselectivity-including completely deteriorating the separation. Interestingly, with increasing isopropylamine concentration, enantioselectivity generally increased for primary amines, decreased for secondary amines, and was approximately the same for tertiary amines.
He then presented work undertaken with Caroline West (University of Orleans) illustrating that the apparent pH of the carbon dioxide mobile phase varied little with isopropylamine concentration (pH was approximately 5–6 irrespective of isopropylamine concentration). David then presented data showing that as the isopropylamine additive concentration increased, more was loaded onto the stationary phase. However, as the organic modifier level was increased (in this case ethanol in the range 7–30% [v/v]), the quantity loaded decreased exponentially. He completed his presentation illustrating how high levels of isopropylamine in the mobile phase can be very useful when scaling up to preparative level.
Claudio Brunelli (Pfizer) continued the theme of SFC in his presentation “From Laboratory to Patient: Implementation of Supercritical Fluid Chromatography in the Pharmaceutical Industry”. He started the presentation by saying that system qualification for GMP use-one of the most recent successes in the SFC evolution-was “a game changer” and was now fully embedded in the entire drug life cycle from discovery to late development for supply to the patient. Claudio noted that SFC is applied at Pfizer for chiral analysis, compounds not stable or not soluble in water, and orthogonal selectivity (to reversed-phase LC). New SFC systems allow methods to be validated and transferred. He exemplified this by the trace analysis of thiourea in a pharmaceutical compound (5).
He described an application for method development of the compound dacomitinib, which is poorly soluble, is labile in aqueous solution, and has a highly complex synthetic route well suited to analysis by SFC. It lability was confirmed by artefacts being observed through peak broadening (analyte interconversion as a result of the addition of water) using UHPLC. The SFC method development involved column screening (unbonded silica, 2-ethyl pyridine, and hybrid silica) with acetonitrile–methanol mix as organic solvent. As there are no in silico SFC retention modelling software packages, an experimental design approach was used using initial cosolvent, methanol–acetonitrile ratio, gradient time, flow rate, temperature, and outlet pressure as factors.
The final method then underwent a technology transfer exercise involving intermediate precision. An observation was made that retention times drifted on a dayâtoâday basis, but this did not cause usability issues with the method. Claudio concluded his presentation by highlighting the application of preparative SFC and chiral method development in his development setting, but commented that column robustness was an area that still required focus.
The next presentation in this session was delivered by Alex Brien (Reach Separations) “Chiral Chromatography-The Challenges and Answers to that Final 5%”, which focused on the difficult 5% of compounds that chiral screens typically don’t resolve. He noted that 95% of compounds can be separated with mobile phases containing an n-alkane–alcohol mixture in conjunction with amylose- and cellulose-based chiral stationary phases. However, the diversity of chemical structures requiring separation is changing with time, requiring Reach to continually modify their screens with more work being successful using SFC (in 2012 to 2014, 45% of separations were completed using SFC, in 2016 this was ~70%) with 95% of hits being identified in under 9 h.
Alex noted that amylose-based chiral columns can offer very different selectivities between vendors and are more susceptible to ageing. He also noted the general impact on peak shape when changing from ammonium hydroxide to DEA and the further enhancements on resolution when increasing the modifier concentration.
Brian Montgomery (HiChrom) standing in for Jeff Caldwell of Princeton Technologies provided a comparison of superficially and fully porous particles for achiral SFC in his presentation “Comparison of Core–Shell & Porous Silica Bonded Phases for SFC”.
In a change of technique, the next presentation was given by James Heaton (Pfizer) titled “Exploring the Practicalities of Hydrophilic Interaction Chromatography (HILIC) in the Pharmaceutical Manufacturing Environment”. After a brief description of the HILIC process (6) and retention mechanisms (ionic, partition, and a mixture of these), he moved onto the effect of ionic strength on peak shape and retention on bare silica phases. A large deterioration in peak shape for the neutral hydroxylated compound (5-hydroxymethyluridine) was observed, the magnitude of ionâexchange varied between different bare silica phases, and acceptable peak shapes for bases at even low buffer salt concentration were all noted when varying the buffer concentration from 1–8 mM for ammonium formate pH 3. Upon changing to a organoâsilane hybrid amide column, a number of differences were noted. These included the poor peak shape for 5-hydroxymethyluridine not being observed, the magnitude of ionâexchange was much less on organoâsilane hybrid HILIC, and there were large increases in retention for neutral compounds on the organo-silane hybrid amide at higher buffer concentrations.
Next James exemplified the impact of salt type on peak efficiency. He noted efficiency for strongly ionogenic compounds was severely compromised using simple acid modifiers like 0.1% (v/v) formic acid (FA) compared to ammonium acetate at the same nominal pH, but the efficiency of neutral compounds and some acids was not compromised. He also noted that low retention for neutral compounds on bare silica was observed compared with amide bonded phase. Very high retention for basic compounds in formic acid was observed because of the absence of NH4+ in the formic acid mobile phase, which increases retention by ion-exchange mechanisms, and retention for pyridine was high because of protonation in the FA mobile phase. On bare silica, he noted increasing ammonium concentration decreased retention for basic compounds. This could be further decreased by substituting ammonium with rubidium cations because Rb+ ions are a stronger ion-exchange displacer than NH4+, as illustrated in the following series: Cs+ > Rb+ > K+ > NH4+ > Na+ > H+ > Li+. These experiments indicated that ion-exchange is not involved in retention of weak bases (adenine–pyridine).
James then discussed the impact of sample diluent and noted peak splitting for some compounds with less than 60% (v/v) acetonitrile in the diluent when injected into a 95% acetonitrile, 5 mM ammonium formate wwpH 3 mobile phase. In addition, increasing injection volume above 1 µL generally had a significant impact on decreasing efficiency (a minimum of 50%) over the range studied, while peak asymmetry was generally maintained.
The next question posed was whether the presence of residual metals in the silica matrix may contribute to peak shape under HILIC conditions. To investigate this EDTA and citrate were investigated as competing complexing agents. EDTA was less useful as an additive because it has poor solubility in acetonitrile–water mixtures >80:20. Catecholamines were analyzed and showed poor efficiency in previously published work on bare silica (7) and are known to coordinate with metals and form complexes. However, no complexation or poor peak shape was observed on the organoâsilane hybrid amide column but there was evidence of complexation on the organo-silane hybrid HILIC column. The addition of 5 mM ammonium citrate did dramatically improve the peak shape on the organo-silane hybrid HILIC column. The addition of small amounts of ammonium citrate (and titrated to pH 3.0 with formic acid) was shown to dramatically improve peak shape for a range of antibiotic molecules too.
Finally, James touched on the issue of reâequilibration. For full system reâequilibration following a HLIIC gradient, approximately 25 column volumes were required and the effect was most pronounced for basic compounds in terms of retention time drift.
The next talk was also on HILIC and was delivered by Alan McKeown (ACT): “A Systematic Approach for Developing Reproducible HILIC Methods”. In the talk, Alan outlined ACT’s recommended method development approach for this important technique.
Alan recommended HLIIC as the optimal approach for molecules with a Log P <0, or if the analyte elutes before caffeine under reversed-phase conditions (8). He recommended this approach over the use of ion-pairing reagents, which are difficult to clean from LC systems once used (and nearly impossible from columns). He highlighted the advantages of HILIC including enhanced mass transfer and lower C-term contribution in the van Deemter equation, improved ionization for MS detection, and lower viscosity of the acetonitrile bulk solvent leading to lower backpressures, which enables the use of higher flow rates, smaller particle sizes, and therefore higher plate count. However, there can be some challenges in using HILIC, including high volume fraction eluents leading to challenges with solubility for hydrophilic analytes, and while retention modelling can be performed for predicting isocratic conditions from gradient models and vice versa, this has been found to be unreliable (unlike reversed-phase LC) (9).
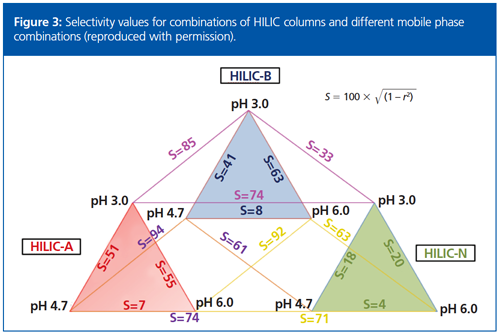
In terms of method development, he noted HILIC stationary phases can be split into acidic, basic, neutral, and “other” or “novel” phases such as zwitterionic and cyclodextrin phases. To maximize selectivity, it is helpful to consider HILIC stationary phases with different characters. To characterize the differences in retention behaviour, Alan used the equation selectivity = 100 × √ (1 – R2), where R2 is the correlation coefficient from plotting the retention of the analytes under two different chromatographic conditions, giving a range of 0 to 1–1 being identical retention conditions and 0 being no retention correlation (10). He went on to discuss how through the analysis of 54 different conditions, three different column types and conditions were identified (columns are HILIC-A [acidic], HILIC-B [basic], and HILIC-N [neutral]) with acetonitrile–10 mM ammonium acetate (pH 3.0, 4.7, and 6.0) gradients (see Figure 3).

This led to a discussion on gradient reâequilibration where a minimum of 50–60 column volumes are initially required for HILIC, but recent work in their laboratory has shown subsequent equilibration requires much less (20 column volumes). Alan stated that for gradient HILIC methods ~10 column volumes are required between injections for reproducible retention times, with the caveat that this was application- and condition-dependent. Like James Heaton, he also discussed the impact of sample diluent noting that this is analyte dependent, but a minimum of 60% (v/v) acetonitrile should be used.
The last presentation of the day was provided by Daniel Esser (YMC) on method scouting and optimization. He outlined a normal-phase method development protocol on immobilized columns where the first-tier mobile phase is an n-hexane–IPA or ethanol gradient plus acidic or basic additives as required. As an alternative to normal phase, Daniel also highlighted a reversed-phase protocol, again based on immobilized columns and a first-tier approach of acetonitrile and formic acid, ammonium acetate, or ammonium bicarbonate gradients depending on analyte properties, for example, 0.1% formic acid to suppress ionization for acidic analytes. The final chiral strategy he outlined was for SFC and again employed immobilized phases with methanol–carbon dioxide mobile phases (again with acidic or basic modifiers as required). He concluded that by using these approaches, chiral method development had a success rate of >90%.
References
- M.D. Jones et al., J. Chromatogr. B200, (2014).
- M. Taraji et al., Anal. Chem. In Press (2017).
- S. Park et al., J. Chromatogr. A In Press (2017).
- W. Ratsameepakai et al., Energy Fuels29, 2485 (2015).
- C. Brunelli et al., Chromatogr. Today (Feb/Mar 2009).
- A.J. Alpert, J. Chromatogr.499, 177 (1990).
- D.V. McCalley, J. Chromatogr. A1193, 85 (2008).
- A.P. McKeown, Chromatogr. Today 8, (Nov/Dec 2015).
- M.R. Euerby, Anal. Bioanal. Chem.407, 9135 (2015),
- U. Neue et al., J. Chromatogr. A1127, 161 (2006).
The programme for Day Three of this illuminating conference will be covered in an upcoming issue of The Column.
E-mail:chromsoc@meetingmakers.co.ukWebsite:www.chromsoc.com




















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

